
Summary
Introduction Drugs inhibiting the mammalian target of rapamycin (mTOR) are approved in the treatment of renal cell carcinoma (RCC), but resistance inevitably emerges. Proposed escape pathways include increased phosphorylation of Akt, which can be down regulated by histone deacetylase (HDAC) inhibitors. We hypothesized that co-treatment with the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat may abrogate resistance in RCC. Methods This phase 1 study evaluated the co-administration of ridaforolimus and vorinostat in patients with advanced solid tumors. The primary objective was to determine the maximum tolerated dose (MTD) in RCC patients. Although all solid tumors were allowed, prior cytotoxic chemotherapy was limited to 1 regimen. Using a modified 3 + 3 dose escalation design, various dose combinations were tested concurrently in separate cohorts. Efficacy was a secondary endpoint. Results Fifteen patients were treated at one of three dose levels, thirteen with RCC (10 clear cell, 3 papillary). Dosing was limited by thrombocytopenia. The MTD was determined to be ridaforolimus 20 mg daily days 1–5 with vorinostat 100 mg BID days 1–3 weekly, however late onset thrombocytopenia led to a lower recommended phase II dose: ridaforolimus 20 mg daily days 1–5 with vorinostat 100 mg daily days 1–3 weekly. Two patients, both with papillary RCC, maintained disease control for 54 and 80 weeks, respectively. Conclusions The combination of ridaforolimus and vorinostat was tolerable at the recommended phase II dose. Two patients with papillary RCC experienced prolonged disease stabilization, thus further study of combined HDAC and mTOR inhibition in this population is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mammalian target of rapamycin (mTOR) kinase is an integral downstream regulator of the phosphoinositide-3 kinase (PI3K)/Akt pathway, a signaling cascade that has been implicated in myriad cellular actions including proliferation, mobility, angiogenesis, and cell survival.[1–3] Altered functioning of this pathway has been linked to tumorigenesis in several human cancers.[2, 4, 5] Inhibition of mTOR directly decreases gene translation, thus reducing protein synthesis and in turn leading to delayed or arrested progression through the cell cycle.[6, 7] Renal cell carcinoma (RCC) has proven to be particularly sensitive to mTOR inhibition,[8, 9] and subsequently two different mTOR inhibitors, temsirolimus and everolimus, have been approved for use as systemic therapy in patients with metastatic RCC based on results from randomized phase III trials.[10, 11]
Another effect of mTOR inhibition involves its role on the downstream transcription of the hypoxia inducible factor-1α (HIF-1α) and its resultant effect on angiogenesis.[9] When it is active, mTOR activation leads to phosphorylation of the 4E-binding protein (4E-BP1) and the S6 kinase (S6K1), which in turn up-regulate HIF-1α. Under hypoxic conditions, the HIF-1α protein translocates into the nucleus to activate gene expression, including vascular endothelial growth factor (VEGF), and stimulate angiogenesis.[12] Normally, HIF-1α is degraded by interaction with the von Hippel-Lindau (vHL) protein complex prior to entering the nucleus, however in many RCC tumor cells a mutated vHL gene leads to HIF-1α accumulation and overexpression.[13, 14] Inhibition of mTOR minimizes HIF-1α production, which serves to temper the enhanced angiogenesis stimulated by HIF-1α in the setting of ineffective, mutated vHL.
Unfortunately, the onset of drug resistance remains a major barrier to prolonged treatment success. Multiple mechanisms have been described that likely contribute to the development of resistance to mTOR inhibition.[15] One potential resistance pathway involves a feedback loop generated in mTOR-inhibited cells that induces up-regulation of Akt phosphorylation and ultimately renders the anti-proliferative effects of mTOR inhibition inadequate to suppress tumor growth.[16] Therefore, a reasonable strategy to avoid or overcome resistance to mTOR inhibitors involves concomitant suppression of phosphorylated Akt (pAkt). HDAC inhibitors block enzymes that return the DNA in histones to a more tightly coiled, less readily transcribed form, resulting in altered transcription patterns of various genes implicated in cell survival, differentiation and proliferation. In a preclinical study by Verheul and colleagues, combining an mTOR inhibitor with a histone deacetylase (HDAC) inhibitor, showed promising activity.[17] HDAC inhibitors have been shown to affect transcription at the DNA level resulting in altered patterns of gene expression implicated in cell survival, differentiation and proliferation.[18] In RCC cell lines, pAKT was predictably upregulated by mTOR inhibition, but with the addition of HDAC inhibition pAKT expression remained at baseline levels. Additionally, HDAC inhibitors are known to inhibit angiogenesis via a HIF-1α mediated process, and the combination treatment revealed further decrease in HIF-1α protein expression, opening the possibility of anti-tumor synergism via dual mechanisms. Figure 1 illustrates the mechanisms of action of both ridaforolimus and vorinostat on the various components of the mTOR pathway. A prior phase I study of the oral HDAC inhibitor vorinostat established a twice daily dosing schedule on three consecutive days every seven, based on pharmacokinetics and toxicity as a single agent, and found this to be safe and tolerable.[19] While not a dose-limiting toxicity in this study, thrombocytopenia appears to be a common class effect of HDAC inhibitor therapy due to effects on megakaryocyte differentiation.[20] A similar phase I dose-finding study investigating the oral mTOR inhibitor ridaforolimus established an optimal dose and schedule of five consecutive days, followed by two days off, based on toxicity and pharmacodynamic endpoints.[21] Based on these promising preclinical findings, we performed a Phase I study with the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat to determine safety and tolerability and investigate whether combination therapy could abrogate drug resistance in RCC.

Mechanism of action of ridaforolimus and vorinostat on the mTOR pathway. Pictorial depiction of the key proteins affected via altered transcription and translation by the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat. Ridaforolimus binds mTOR at the rapamycin binding domain resulting in allosteric kinase inhibiton and leading to downregulation of various cell cycle regulators as well as HIF-1. Vorinostat exerts its effects on histone deacetylation to decrease transcription, culminating in decreased pAkt and HIF-1. Note: PDK phosphoinositide-dependent protein kinase 1, mTOR mammalian target of rapamycin, S6K1 S6 kinase 1, IRS insulin receptor substrate, eIF eukaryotic initiation factor, HIF hypoxia inducible factor
Patients and methods
Patients
Eligible patients were initially considered for the study if they had metastatic RCC refractory to prior systemic therapy. However due to emerging data suggesting efficacy of this approach in other tumor types,[22–24] the protocol was amended to include patients with all solid tumors and lymphomas. Enrollment was limited to patients whose disease was refractory to at least one line of therapy but who had received no more than one prior cytotoxic chemotherapy. There was no limit on the number of prior targeted or immunotherapies. Patients were > 18 years of age, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had adequate organ function, and measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Female patients of childbearing age were required to have a negative serum or urine pregnancy test ≤ 5 days prior to study registration and agree to use an effective method of contraception. Restrictions on timing of prior therapies included cytotoxic chemotherapy, radiotherapy or immunotherapy ≤ 3 weeks before starting treatment, the use of bevacizumab ≤ 6 weeks prior to day 1 of treatment, or a lack of recovery from previous treatment-related acute adverse events (AEs). For patients who had received prior targeted therapies, treatment must have been discontinued at least five half-lives prior to initiation of study treatment. Key exclusion criteria included an active infection requiring antibiotics, uncontrolled diabetes or hyperlipidemia, and a known diagnosis of HIV. Stable brain metastases were permitted. All patients were required to sign written informed consent. The protocol was approved by the institutional review board of Fox Chase Cancer Center and Merck. The study followed the Declaration of Helsinki and good clinical practice guidelines.
Study design
This was a single institution, phase I investigator initiated study with a modified 3 + 3 dose escalation design. The primary objectives of the study were to characterize the safety and tolerability of the dose combination of vorinostat and ridaforolimus as well as determine a maximally-tolerated dose (MTD). Secondary objectives included assessments of response rate (RR), progression free survival (PFS), and overall survival (OS) in all patients as well as in the cohort of RCC patients only. Dose escalation among six candidate dose combinations was planned, with the design allowing for concurrent testing of multiple dose levels by escalating each drug in separate cohorts (Fig. 2a and b). Predefined criteria for dose escalation needed to be met prior to opening the next group of doses to patient accrual. This novel concurrent dose escalation design was previously reported by Huang and colleagues.[25] Both drugs were available in oral formulations. Ridaforolimus was self-administered at the assigned dose level daily for five days, followed by two days off of drug, repeating every week of a 21-day cycle. Vorinostat was dosed once or twice daily for three days on, four days off, repeating weekly on the same 21-day cycle (Fig. 2c). Treatment was continued at the defined dose level until disease progression, unacceptable toxicity or adverse events as determined by either the investigator or the patient, or withdrawal from study. Any drug-related grade 3 or 4 toxicity occurring during the first 3 weeks of treatment (except nausea, vomiting, diarrhea, serum lipid elevation, or transient electrolyte abnormality that resolved to a grade of 0–2 with medical management) was considered a dose limiting toxicity (DLT). Cohort expansion could proceed to the next group of dose levels if no DLTs were reported in the first 3 patients treated. If one DLT was recorded, cohort expansion to six patients was required at that dose level, and further dose escalation was suspended until all patients could be assessed. Dose escalation was halted if two or more of any DLT was recorded, and further accrual to other dose levels was determined by predefined specifications. The dose escalation protocol was followed to determination of a MTD, denoted as the highest dose at which no more than one of six patients experienced a DLT, and expanded to a total of 12 patients.

Trial Design Including Dose Escalation and Treatment Schedule a Composite dose level combinations of ridaforolimus and vorinostat, with the starting dose level emphasized in bold text. b Parallel dose trial design allowing for separate escalation and de-escalation of each individual drug in patient cohorts based on toxicity assessments to determine optimal dosing combinations. c Treatment schedule for combination oral administration. Vorinostat was dosed three days on followed by 4 days off starting on day one of each cycle, with daily or twice daily dosing dependent on the dose level. Ridaforolimus was given daily for 5 days with two off days. A cycle was considered to be 21 days long. Baseline imaging and initial restaging scans were assessed as marked. Note: mg milligrams, BID twice daily, C1 cycle 1
Procedures
Baseline evaluations consisted of a physical examination, complete blood count, complete metabolic panel, coagulation studies, fasting lipids, electrocardiogram, computed tomography or magnetic resonance imaging of the chest, abdomen, pelvis, and bone scan. Safety evaluations were also performed at baseline, at 2 weeks, then weekly for 4 weeks. Thereafter, evaluations were performed on the first day of each subsequent cycle. Toxicity noted in patients receiving ridaforolimus and vorinostat were graded using the National Cancer Institute common toxicity criteria for adverse events (CTCAE) version 4.0 grading system. Radiographic restaging occurred after every two cycles for the first six cycles and then could be extended to every four cycles per physician preference. Evaluation of response followed the RECIST v. 1.1 guidelines.
Data analyses
Determination of DLTs and MTD is outlined above. Kaplan Meier curves were used to estimate PFS and OS. Proportions and 95 % CIs were used to evaluate the treatment completion rate. Results are reported as of data cutoff on May 1, 2014.
Results
Patient characteristics
Fifteen patients were treated at one of three dose levels. Thirteen patients (87 %) had RCC, three of which had papillary RCC and ten that had clear cell RCC (ccRCC). There was one patient with esophageal carcinoma and one with a small bowel carcinoid tumor. The median age was 66 years (range: 57–80 years). All patients had a performance status of 0–1 at study initiation and had progressed on prior systemic therapy. Seven patients (47 %) had received only one prior therapy, while five patients (33 %) and three patients (20 %) had received two or greater than or equal to three prior regimens, respectively. More than half of the patients (8, 53 %) had been treated previously with an mTOR inhibitor, including the patient with the carcinoid tumor. Table 1 displays the overall patient characteristics. At the time of data cut-off, no patients remained on study.
Safety and tolerability
Dose escalation progressed from dose level #1 to the parallel dose levels denoted as #2 and #3 after no DLTs were seen at the initial dose level, as per protocol parameters (Fig. 2a and b). Inability to complete 80 % of doses during cycle 1 due to persistent grade 1–2 thrombocytopenia led to two DLTs among six patients at dose level two. There were no DLTs among the first three patients treated at dose level three, thus triggering an exploratory expansion to six patients at this level. However, five of the six patients ultimately required dose reduction due to thrombocytopenia, and thus this dose combination was determined intolerable for prolonged use due to thrombocytopenia and equivalent to a DLT. Thus, dose level #1 (ridaforolimus 20 mg daily, vorinostat 100 mg BID) was determined to be the MTD. There were no other DLTs. During subsequent treatment, further dose reduction (to ridaforolimus 10 or 20 mg daily with vorinostat 100 mg daily) was necessitated in all patients who received >2 cycles (7 patients), with grade 1–2 thrombocytopenia implicated in 6/7 cases. The incidence of thrombocytopenia appeared to be directly related to vorinostat dosing. Ultimately, after review of both cycle 1 and long term toxicity and efficacy data, ridaforolimus 20 mg daily X 5 (days 1–5) with vorinostat 100 mg daily X 3 (days 1–3) weekly was determined to be the recommended phase two dose (RP2D). Notably, the reduction of vorinostat from twice daily (the MTD) to once daily in the RP2D was chosen to minimize thrombocytopenia.
The most common AEs that developed in two or more patients are presented in Fig. 3. AEs of all grades found in greater than 50 % of patients included oral mucositis (12 patients, 80.0 %), fatigue (11 patients, 73.3 %), anorexia (11 patients, 73.3 %), thrombocytopenia (11 patients, 73.3 %), hyperglycemia (9 patients, 60.0 %), and anemia (9 patients, 60.0 %). The only grade 3 AEs occurring in more than one patient were anemia and hyperglycemia (4 patients and 2 patients, respectively). Grade 3 AEs occurring in one patient only included diarrhea, fatigue, and mucositis. There were no grade 4–5 toxicities. Six patients (40 %) discontinued the study due to toxicity.

Most Common Drug-Related Adverse Events Chart depicting the most commonly reported adverse events by patients. Individual toxicities are graphed by number of patients reporting that side effect and are color coded by grade according to the Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0. Note: ALT alanine transaminase, AST aspartate transaminase
Efficacy
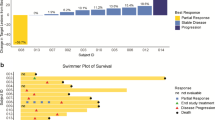
A swimmer’s plot illustrating PFS and OS for all 15 patients can be found in Fig. 4. Four patients, all with RCC, maintained stable disease for 19, 21, 54 and 80 weeks, respectively. Three of these patients had progressed on prior mTOR inhibitor therapy, including the two patients with the longest periods of disease control, both of whom had papillary RCC. The patient who was treated on study for 54 weeks had previously been treated with the mTOR inhibitor temsirolimus and had sustained a prolonged response lasting 27 months with that agent. Both of these patients with papillary RCC and sustained duration of treatment with the study regimen were still alive at the time of data cut-off. No patients achieved an objective response by RECIST criteria.

Swimmer’s Plot of Progression Free and Overall Survival Chart depicts progression free and overall survival of each individual patient as a function of time as measured in weeks. Patients are divided with those having received a prior mTOR inhibitor on the top and those without prior mTOR inhibition towards the bottom. Symbols indicate whether patients were still living at the time of data cut-off; no patients remained on study. Note: mTOR mammalian target of rapamycin, PFS progression free survival, OS overall survival, Wks weeks
The median PFS for all 15 patients treated on protocol was 17.1 weeks (95 % CI: 6.6–21.4 weeks). In this cohort of patients where the majority had been treated with two or more prior regimens in the metastatic setting, 9/15 (60 %) had a PFS greater than 16 weeks. All nine of these patients had RCC. The median OS was 40.7 weeks (95 % CI: 28.3 weeks-not reached). Figure 5 shows the Kaplan Meier curves for both PFS and OS. At the time of data cut-off, five patients (33 %) were still alive, but no patients remained on study.

Kaplan Meier Plots of Progression free and Overall Survival a Kaplan Meier curve of progression free survival with 95 % confidence bounds depicted as dotted lines. b Kaplan Meier curve of overall survival with 95 % confidence bounds depicted as dotted lines
Discussion
The goal of our study was to explore the utilization and feasibility of dual inhibition of mTOR and HDAC in an attempt to abrogate a primary mechanism of resistance via pAKT, as well as via synergistic inhibition of HIF-1α, in patients with metastatic RCC. Thrombocytopenia was the primary DLT with this drug combination, and it appeared to be both dose-related and to increase in severity with repeated dosing. This was not unexpected based on previous experience and reports with HDAC inhibitors, particularly vorinostat.[19, 26] However, owing to the 3x3 dosing design of the study, a tolerable dosing combination of ridaforolimus 20 mg daily with vorinostat 100 mg daily was deemed acceptable for further evaluation in a subsequent phase II trial. This combination did not lead to any other DLTs, most other toxicities were manageable and completely reversible, and no grade 4 or 5 toxicities were reported.
While this study was not designed to assess efficacy, it is encouraging that a subset of patients did appear to derive benefit from the drug combination. Additionally, while there were no objective tumor responses reported, two patients achieved a prolonged period of disease stability while on treatment. Interestingly, both of these patients had papillary RCC histology and both had progressed on prior mTOR inhibitor therapy (each had received temsirolimus as a first line agent). Notwithstanding the inherent statistical limitations of applying the findings in two patients of a relatively small cohort, these results warrant further investigation of this drug combination in patients with metastatic papillary RCC.
Due to the relative rarity of non-clear cell RCC, strong data supporting the efficacy of any single agent in these tumors is lacking. Arguably the best published data from a phase III trial setting comes from a retrospective analysis of the global advanced renal cell carcinoma (ARCC) trial.[10, 27] The original trial compared temsirolimus versus interferon-α (IFN-α) versus the combination in patients with metastatic RCC and poor prognostic features. The results showed an OS benefit for the patients on the temsirolimus alone arm and led to the approval of temsirolimus for use in this population. Because 20 % of the patients in this trial harbored non-clear cell histology, predominantly papillary, a subsequent retrospective subset analysis was performed and reported an OS of 11.6 months in the temsirolimus alone arm versus 4.3 months with IFN-α alone in the patients with non-clear cell RCC. The 10 patients with pure papillary RCC only were reported to have an OS with temsirolimus of 13.2 months. Though this data is retrospective, and pathology was not centrally reviewed, it marks the only reported phase III data supporting a specific therapeutic benefit of mTOR inhibition for patients with non-clear cell histology.
Though this trial did not set out to validate a role for combined mTOR/HDAC blockade specifically in patients with papillary RCC, recent work published by Chaux and colleagues lends pre-clinical support to this potential strategy.[28] In their study the investigators examined a variety of biomarkers from the nephrectomy specimens of 54 patients with papillary RCC. They noted significant increases in the expression of 4E-BP1 (which is activated by mTOR) and HIF-1α in the tumor samples as compared to normal kidney tissue specimens. Consequently, the authors concluded that their results support a scientific rationale of dual targeting of mTOR and hypoxia-induced pathways in papillary RCC.
Our study establishes a reasonably safe and tolerable dosing regimen for the combination of ridaforolimus and vorinostat and preliminary evidence of efficacy in treating patients with metastatic RCC. The promising results in two patients with papillary RCC are intriguing, and in light of supporting pre-clinical data, warrant further study of the combination of mTOR and HDAC inhibition in a proof of concept phase II trial.
References
Chan S (2004) Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer 91(8):1420–1424
Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441(7092):424–430
Schmelzle T, Hall MN (2000) TOR, a central controller of cell growth. Cell 103(2):253–262
Robb VA et al (2007) Activation of the mTOR signaling pathway in renal clear cell carcinoma. J Urol 177(1):346–352
Mills GB et al (2001) The role of genetic abnormalities of PTEN and the phosphatidylinositol 3-kinase pathway in breast and ovarian tumorigenesis, prognosis, and therapy. Semin Oncol 28(5 Suppl 16):125–141
Barbet NC et al (1996) TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell 7(1):25–42
Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18(16):1926–1945
Pantuck AJ et al (2003) Pathobiology, prognosis, and targeted therapy for renal cell carcinoma: exploiting the hypoxia-induced pathway. Clin Cancer Res 9(13):4641–4652
Thomas GV et al (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 12(1):122–127
Hudes G et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281
Motzer RJ et al (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372(9637):449–456
Treins C et al (2002) Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem 277(31):27975–27981
Raval RR et al (2005) Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 25(13):5675–5686
Clifford SC et al (1998) Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: Evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosom Cancer 22(3):200–209
Hudes GR (2009) Targeting mTOR in renal cell carcinoma. Cancer 115(S10):2313–2320
O’Reilly KE et al (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66(3):1500–1508
Verheul HM et al (2008) Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin Cancer Res 14(11):3589–3597
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5(9):769–784
Kelly WK et al (2005) Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol 23(17):3923–3931
Matsuoka H et al (2007) Mechanisms of HDAC inhibitor-induced thrombocytopenia. Eur J Pharmacol 571(2):88–96
Mita MM et al (2008) Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol 26(3):361–367
Gupta M et al (2009) Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood 114(14):2926–2935
Morgan S, Cranmer L Effect of the combination of mTOR inhibitor ridaforolimus and HDAC inhibitor vorinostat on in vitro synergism in synovial sarcoma, osteosarcoma, and a range of other tumor subtypes. In Journal Of Clinical Oncology. 2011. Amer Soc Clinical Oncology 2318 mill road, STE 800, Alexandria, VA 22314 USA.
Wedel S et al (2011) Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci 88(9):418–424
Huang X et al (2007) A parallel phase I/II clinical trial design for combination therapies. Biometrics 63(2):429–436
Confidential Investigators Brochure: Vorinostat, I. Merck and Co., Editor
Dutcher JP et al (2009) Effect of temsirolimus versus interferon-alpha on outcome of patients with advanced renal cell carcinoma of different tumor histologies. Med Oncol 26(2):202–209
Chaux A et al (2012) Immunoexpression status and prognostic value of mammalian target of rapamycin and hypoxia-induced pathway members in papillary cell renal cell carcinomas. Hum Pathol 43(12):2129–2137
Acknowledgments
The authors would like to thank the patients who enrolled in this trial, as well as their families
Funding Sources
This work was supported in part by Merck Inc.
Financial Disclosures
ERP has served as a consultant for Merck Inc.
Precis for use in the Table of Contents
HDAC inhibition may abrogate Akt mediated resistance to mTOR inhibition. When combined in this phase I trial, the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat were safe in combination and preliminary efficacy results suggest proof of concept.
Author Contributions
Conception and Design: ERP
Acquisition of data: YNW, LM, AJO, CSD, SKR, CHT, ERP.
Analysis and interpretation of data: MZ, KD, and ERP.
Writing, review and/or revision of the manuscript: All authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial registration ID: NCT01169532
Neither the submitted manuscript nor any similar manuscript, in whole or in part, other than an abstract, is under consideration, in press, published, or reported elsewhere.
Rights and permissions
About this article

Cite this article
Zibelman, M., Wong, YN., Devarajan, K. et al. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest New Drugs 33, 1040–1047 (2015). https://doi.org/10.1007/s10637-015-0261-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0261-3