
Summary
Inhibition of heat shock 90 (Hsp90) molecular chaperones allows targeting of multiple proteins involved in tumorigenesis. We investigated the safety, recommended phase 2 dose (RP2D), and pharmacokinetic and pharmacodynamic profile of onalespib (AT13387), a potent synthetic Hsp90 inhibitor, administered on days 1, 2, 8, 9, 15, and 16 of 28 day cycles (QDx2/week) in a phase I trial. This study followed an accelerated titration design with a starting dose of 20 mg/m2/dose and a standard 3 + 3 dose escalation design for dose level 4 (120 mg/m2/dose) and above. Additional patients were enrolled at the RP2D with mandatory paired tumor biopsies to assess modulation of 210 client proteins using reverse phase protein array analysis. Thirty-one patients were treated; RP2D was established at 160 mg/m2/dose on the QDx2/week schedule. Common toxicities were gastrointestinal, hepatic, and hematologic. Pharmacokinetic profile was linear and plasma levels increased proportionally with dose (T½ ~8 h). No responses were observed; eight patients had stable disease for > 2 cycles with one patient remaining on study for 6 cycles. Target engagement was demonstrated by transcriptional upregulation of Hsp70 and Hsp27 in PBMCs. Statistically significant modulation of client proteins was not achieved in the 9 paired tumor biopsies evaluated; however, hierarchical clustering revealed two subgroups of patients with differential patterns of protein expression. Further combination studies are needed in order to target prospective driver oncoproteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 90 kDa heat shock protein (Hsp90) is an evolutionarily conserved class of molecular chaperone proteins that exert housekeeping functions within cells. These proteins assist in the folding, stabilization, activation, and proteolytic turnover of newly synthesized proteins, called “client proteins” [1]. Among these client proteins are those which are involved in aberrant cellular functions responsible for the hallmarks of cancer, including angiogenesis (VEGF, VEGFR, HIF-1), growth factor independence (RAF, EGFR, HER2), resistance to anti-growth signals (CDK4), tissue invasion and metastases (MET, MMP2), and avoidance of apoptosis (AKT, RIP, Survivin, Bcl-2) [2]. The protection afforded to aberrant expression of these proteins by the Hsp90 molecular chaperone allows for the growth and survival of cancer cells. Inhibition of Hsp90 leads to degradation of these aberrant proteins through the ubiquitin-proteosome pathway, allowing for simultaneous targeting of multiple pathways involved in the potentiation of cancer cells, forming the basis for inhibition of Hsp90 as an attractive anti-cancer therapy [3, 4].
Previous development of Hsp90 inhibitors, including geldanamycin and its analogues (17-AAG, 17-DMAG, and IPI-504), was limited by heptotoxicity and unstable formulations with limited clinical activity [5–7]. Onalespib (AT13387) is a second-generation, non-ansamycin, small molecular inhibitor of Hsp90, with a Kd of 0.71 nM. It acts by binding at the N-terminal ATP binding site of Hsp90, leading to the inhibition of chaperone function and promotion of the degradation of its client proteins. Although intermittent, twice-a-week dosing of onalespib was the chosen schedule for the initial clinical study in humans [8], alternative dosing schedules with daily administration for the first 2 days per week (QDx2/week) demonstrated consistently greater tumor growth inhibition and intratumoral drug accumulation in several human xenograft models [9]. Such a schedule of dosing allowed for increased drug exposures in tumor while drug was cleared from the plasma, potentially improving the therapeutic window with sustained client protein depletion and minimizing damage to normal tissues. We conducted a phase 1 trial of onalespib administered QDx2/week for 3 out of 4 weeks to establish its safety, recommended phase 2 dose (RP2D), and pharmacokinetic profile. Paired tumor biopsies were obtained from patients enrolled on the expansion phase at the RP2D to assess client protein modulation.
Patients and methods
Patient population
Patients 18 years and older with histologically confirmed advanced solid tumors that had progressed or recurred after at least one line of chemotherapy were eligible. Patients were required to have an Eastern Cooperative Group performance status of 0–2 and adequate organ function, defined as absolute neutrophil count ≥ 1500/μL, platelet count ≥ 100,000/μL, total bilirubin ≤ 1.5X institutional upper limit of normal (ULN), AST and ALT ≤ 2.5X ULN, and creatinine < 1.5X ULN. Patients enrolled on the expansion cohort at the maximum tolerated dose (MTD) were required to have disease amenable to biopsy and be willing to undergo paired tumor biopsies. Previous treatment must have been completed at least 4 weeks prior to enrollment. Exclusion criteria included uncontrolled intercurrent illness, unstable brain metastases, gastrointestinal bleeding within 1 week of starting treatment, and prolonged QTc (defined as QTc > 450 msec for men and 470 msec for women). This trial was conducted under a National Cancer Institute (NCI)-sponsored IND with institutional review board approval. Informed consent was obtained by the investigators from each participant, and protocol design and conduct followed all applicable regulations, guidances, and local policies (ClinicalTrials.gov Identifier: NCT01246102).
Study design
This was an open-label, phase 1 trial of single-agent onalespib in patients with advanced solid tumors. Onalespib was supplied by the Division of Cancer Treatment and Diagnosis, NCI, under a collaborative research and development agreement with Astex Pharmaceuticals. Dosing schema for this study was based on preliminary toxicity data available from study NCT00878423, where intermittent twice weekly dosing was tested (days 1, 4, 8, 11, 15, and 18 in 28 day cycles) [8]. Toxicity data from this previous study determined 120 mg/m2/dose to be the maximum-tolerated-dose based on the incidence of moderate toxicities: 10 of 13 patients at this dose experienced visual disturbances, including one incidence of grade 3 visual disturbance.
In our study, onalespib was administered on 2 consecutive days each week for 3 weeks of a 4-week cycle (days 1, 2, 8, 9, 15, and 16). Treatment was repeated every 4 weeks, until disease progression or the development of intolerable toxicities. We followed an accelerated titration design [10], with initial dose level increases in 100 % increments starting at 20 mg/m2/dose until dose level 4 (120 mg/m2/dose), based on the MTD established for the twice weekly schedule. At dose level 4 and above, patients were enrolled following a standard 3 + 3 design.
Toxicities were graded according to the Common Terminology Criteria for Adverse Events (CTCAE version 4.0). Dose limiting toxicity (DLT) was defined as grade 4 neutropenia > 5 days or febrile neutropenia, grade 4 thrombocytopenia, and grade 3–4 non-hematologic toxicities felt to be related to study drug and occurring during the first cycle. Grade ≥ 3 nausea, vomiting, or diarrhea, or occurrence of bloody diarrhea was considered dose-limiting if refractory to symptomatic management. Grade 2 or higher ocular toxicity that did not resolve to grade 1 or less within 2 weeks of withholding treatment was considered dose-limiting. Treatment was held for grade 3 hematologic toxicities (other than lymphopenia, which was not considered dose modifying) until recovery to ≤ grade 2 prior to restarting at the same dose level; treatment was reinitiated at the next lower dose level for grade 4 hematologic toxicities and grade 3 thrombocytopenia. Treatment was held for grade 3 non-hematologic toxicities until recovery to ≤ grade 2 (≤ grade 1 for ocular toxicities) prior to re-initiating treatment at the next lower dose level.
The maximum tolerated dose was defined as the dose level at which no more than 1 of 6 evaluable patients experienced a DLT during the first cycle of treatment. At the MTD, 12 additional patients were enrolled in the expansion cohort with protocol-mandated tumor biopsies prior to drug administration and within 24 h after the last dose of drug during cycle 1 (on day 16 or 17). Radiologic response assessments by computerized tomography scans were performed at baseline and every 2 cycles. Tumor response was evaluated according to the Response Evaluation Criteria in Solid Tumors (RECIST version 1.1).
Safety assessments
History and physical examination and laboratory evaluations (complete blood count and serum chemistries) were performed prior to each treatment week. Electrocardiogram was performed prior to the start of each cycle, and as clinically indicated. Ophthalmologic exam (including fundoscopic exam, visual acuity, intraocular pressure, assessment of visual fields and measurement of color vision, and ocular coherence tomography as indicated) was performed prior to enrollment, after every third cycle, and as clinically indicated thereafter.
Pharmacokinetic analyses
Serial plasma samples of onalespib were collected prior to drug administration and at 0.5, 1, 1.5, 2, 3, 4, 7, 10, 22, and 24 h after the start of infusion on day 1 and day 15 of the first cycle. Urine samples for pharmacokinetic analyses were collected at baseline, and for 24 h after the first drug administration. Reference samples of onalespib (NSC749712) and its deuterated analog, used as the internal standard for the assay, were obtained from the Developmental Therapeutics Program, NCI (Bethesda, MD). After protein precipitation, plasma or urine samples were analyzed by reverse-phase high performance liquid chromatography using positive-ion electrospray ionization with selected reaction monitoring mass spectrometric detection. Standard curves (from 0.004 to 4.0 μM) were prepared and run together with patient plasma or urine samples.
Pharmacodynamic analyses
Paired tumor biopsies (18 gauge core needle biopsies, 2 cores) were obtained from one patient during the dose escalation phase (dose level 3) for assessment of Hsp70 and Hsp27 transcriptional regulation by RT-PCR. Peripheral blood mononuclear cells (PBMCs) were obtained prior to drug administration and prior to dosing on days 2, 15, and 16 of cycle 1 from 9 patients at dose levels 1 through 5 to measure Hsp70 protein levels by Western blot as described previously [11]. Blood samples were collected prior to administration of drug and prior to dosing on day 16 of cycle 1 for serum analysis of Hsp70 and two markers of apoptosis (full-length cytokeratin 18 [M65] and caspace-cleaved cytokeratin 18 [M30]) by M30 Apoptosense and M65 ELISA kits (PEVIVA AB).
Paired tumor biopsies were required during the expansion phase of the study for evaluation of Hsp90 client protein modulation by reverse phase protein array (RPPA) analysis, performed by the Functional Proteomics core facility at the University of Texas MD Anderson Cancer Center, as previously described [12]. Wilcoxon signed-rank test was applied to 210 client protein expression levels for analysis of change in levels following treatment. For each protein, the within patient difference in expression was computed and the median change in expression compared to zero. To cluster patients into sub-groups with similar patterns of basal and treatment-induced changes in expression among all 210 proteins, we applied the hierarchical clustering method using Euclidean distance and complete linkage for the log2 expression changes. Wilcoxon rank sum test was used to identify proteins differentially altered between patient subgroups. All statistical tests were adjusted for multiple testing using the Benjamini and Hochberg procedure.
Results
Patient population and disposition
Thirty-one patients were enrolled and treated in this study between November 2010 and December 2013, including a 12 patient expansion cohort at the MTD for evaluation of pharmacodynamic endpoints. Three patients in the expansion cohort were inevaluable for pharmacodynamic endpoints, one due to disease progression after the first week of treatment requiring palliative radiation, the second due to development of a vascular thrombus requiring thrombolytic therapy, precluding our ability to pursue a second biopsy, and the third due to insufficient tumor material available from the paired biopsies. Median age was 55 (range 36–78), male-to-female ratio was 17:14, and the mean number of prior treatments was 4 (range 2–12). Additional patient demographics are shown in Table 1. No responses were observed; eight patients had stable disease at the first restaging after 2 cycles and one patient with hepatocellular carcinoma remained on study for 6 cycles.
Adverse event profile
As shown in Table 2, the most frequent adverse events were hematologic and gastrointestinal. Twelve patients on study experienced some form of visual disturbance, occurring at doses of 120 mg/m2/dose and above. Symptoms were transient, included intermittent blurry vision, visual flashes, and changes in light/dark accommodation, and were of grade 1 or 2 severity. No objective findings were detected on ophthalmologic exam, and symptoms did not persist for more than one day in any case. Nine patients experienced a grade 1 rash, most commonly of acneiform type. Six patients experienced grade 1 QTc prolongation, though no more than 10–15 msec above baseline and with no changes in morphology. Two patients had mild infusion reactions, described as a “warmth” or “flushing” sensation, that resolved within a few hours after completion of the infusion. One patient experienced a DLT at dose level 5 with persistent grade 3 liver enzyme abnormalities despite interrupting treatment for longer than 2 weeks, and two patients experienced DLT at dose level 6 with gastrointestinal hemorrhage during the first week of treatment (one grade 1 and one grade 2 hemorrhage). Preclinical toxicology studies in rabbits had revealed cecal hemorrhage as a dose-related toxicity. Following review of the preclinical data and the patients’ medical status, the decision was made to take the patients off study. Based on the overall tolerability of the regimen, the dose of 160 mg/m2/dose was determined to be the RP2D.
Pharmacokinetic analyses
Over the range of doses from 20–210 mg/m2, plasma exposure to drug (AUC) was proportional to dose (Fig. 1). There was no significant plasma accumulation of drug with repeated dosing. Plasma half-life was ~8 h, and < 10 % urinary drug excretion was detectable for 24 h after drug administration.

Pharmacokinetics of onalespib showed a dose-proportional increase in AUC from doses ranging from 20 to 210 mg/m2/dose
Pharmacodynamic studies
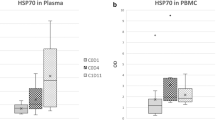
Analysis of pharmacodynamic markers in a subset of patients during the dose escalation phase confirmed on-target activity of onalespib. One patient at dose level 3 (80 mg/m2/dose) underwent optional paired tumor biopsies, which demonstrated indirect evidence of target engagement with transcriptional upregulation of Hsp70 and Hsp27 mRNA, > 1.5-fold and > 2-fold, respectively, following treatment (Fig. 2a). Additionally, evidence of increased Hsp70 protein expression was observed in PBMC samples from patients at dose levels 2 through 5 (RP2D) (Fig. 2b). Serum markers of cellular apoptosis also increased in 5 of 6 patients at dose levels 1 through 4, although the change was not significant (data not shown).

a Paired tumor biopsies obtained from one patient at dose level 3 prior to drug administration and after the last dose of treatment during the first cycle on day 17. Greater than 1.5-fold induction of Hsp70 mRNA and greater than 2-fold induction of Hsp27 mRNA was detected. b Blood samples for PBMCs were collected from 9 patients at dose level 1 through 5 (RP2D) prior to drug administration cycle 1 day 1, 2, 15, and 16. Increased Hsp70 protein expression was measured by Western blot in PBMCs from eight patients at dose level 2 through 5, with recovery to near-baseline levels observed prior to the next week of dosing. Only Patient #1 at dose level 1 showed no increase in Hsp70 expression
Twelve patients were enrolled in the expansion cohort, and 9 were evaluable for pharmacodynamic endpoint analyses with paired tumor biopsies obtained at baseline and after the last dose of drug during the first cycle (either cycle 1 day 16 or 17). Analysis of within-patient protein expression changes for each of the 210 selected client proteins measured by RPPA showed no proteins with statistically significant changes across all 9 patients. Pretreatment protein expression levels were highly heterogeneous between the patients (Fig. 3a). We calculated the difference in log2 normalized expression levels from pre- to post-treatment and performed a hierarchical clustering analysis to look for subgroup effects (Fig. 3b). We found that the expression pattern of patient #23, who had a protein loading correction factor greater than our cutoff of 2.5, was different from the other patients and needed to be excluded from further statistical analysis. The hierarchical clustering result showed two subgroups with different expression patterns among all 210 proteins (subgroup 1: patients 21, 22, 31; subgroup 2: patients 28, 24, 30, 27 and 29). Wilcoxon rank sum test was applied to evaluate which proteins might explain the changes in expression values between these two groups. Table 3 shows the top ten proteins with smallest adjusted p-values for differential patterns after treatment between the two subgroups. For example, expression of CHK2 was increased and SERPINE1 was decreased in subgroup 1, while the levels of expression did not change following treatment in subgroup 2. Additional analysis of 35 client proteins for which phosphorylation state was assessed in the RPPA panel demonstrated a > 2-fold decrease in CHK1 and CHK2 phosphorylation after treatment in subgroup 1 only.


RPPA expression analysis of 210 Hsp90 client proteins from 9 patients before and after onalespib treatment. a Heatmap of log2 normalized pre-treatment protein expression levels showed significant heterogeneity. b Heatmap of log2 normalized expression differences between pre- and post-treatment with hierarchical clustering identified one patient (patient #23) as separate from the others, and two main patient groups: subgroup 1 of patients #31, 22, and 21, and subgroup 2 of patients #29, 27, 30, 24, and 28
Conclusions
This phase 1 study sought to evaluate the safety of the alternate dosing schedule of QDx2/week for 3 out of 4 weeks, and established the RP2D of 160 mg/m2/dose. Exposure increased proportionally with increasing dose, with a half-life ~8 h. The results for plasma drug exposure and half-life, as well as urinary excretion, confirm the work of Shapiro et al. [8]. We observed no responses, but 8 patients had stabilization of disease at the first restaging, and one patient with hepatocellular carcinoma remained on study for 6 cycles. We observed indirect evidence of target engagement with transcriptional upregulation of Hsp70 and Hsp27, and increased Hsp70 protein expression in PBMCs at dose levels 2 through 5 (the RP2D). This is the first report to extensively evaluate changes in the expression levels of 210 client proteins in paired tumor biopsies from patients treated with onalespib. Statistically significant modulation of client proteins was not demonstrated across the 9 patients; however, two expression subgroups were identified by hierarchical clustering. The pre- to post-treatment change in expression of 10 proteins, including Raf-1, c-Src, CHK2, serpin E1, STAT5A, eEF-2 kinase, STAT3, RAB11A/B, MAP2K2, and PRAS, approached statistical significance when compared between the two subgroups. Since objective clinical responses were not observed in this study, the clinical significance of these changes could not be established. Possible explanations for this less than robust response in client protein modulation may involve compensatory induction of the heat shock response through increased expression of heat shock factor 1 (HSF1), Hsp70, and Hsp27, which have been implicated in attenuation of response to Hsp90 inhibition and anti-apoptotic response in cancer cells [13, 14]. There likely is a differential effect on client proteins across tumor types. The variety of tumor types present in the patients who underwent paired tumor biopsies may have contributed to the lack of consistent effects across the biopsies evaluated; analysis of multiple samples of the same tumor or of multiple tumors with the same genetic background might improve the ability to detect changes in relevant client proteins. It is possible that this schedule of drug administration did not result in sufficient tumor exposure to cause significant modulation of client proteins, contributing to the lack of clinical response observed. Alternatively, it has also been proposed that advanced tumors are inherently refractory to efficient Hsp90 inhibition alone due to the development of compensatory resistance pathways. Emerging evidence indicates that Hsp90 inhibitors may play a role in preventing the emergence of resistance, supporting development of this agent in combination with other therapies to increase the effectiveness of earlier lines of therapy [15].
Hsp90 inhibitors have been evaluated on multiple schedules with the aim of maximizing intratumoral drug exposure to allow for sustained client protein depletion while minimizing collateral damage to normal tissues. While this particular dosing schedule showed an acceptable safety profile, we were unable to demonstrate significant depletion of the levels of client proteins. This study illustrates the complexity of defining the optimal dosing schedule for this agent in particular, and Hsp90 inhibitors as a class. Since Hsp90 inhibitors affect multiple proteins with downstream consequences, the specific protein and the degree of inhibition required needs to be defined within the context of a particular tumor type to be able to pick the biomarker of choice. Further understanding of the particular client protein(s) driving progression of disease and the proteins involved in parallel signaling pathways that may be contributing to resistance is needed to provide additional avenues to maximize therapeutic benefit in combination studies.
References
Pratt WB (1998) The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med 217(4):420–434
Sidera K, Patsavoudi E (2008) Extracellular HSP90: conquering the cell surface. Cell Cycle 7(11):1564–1568
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5(10):761–772. doi:10.1038/nrc1716
Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10(8):537–549. doi:10.1038/nrc2887
Neckers L, Schulte TW, Mimnaugh E (1999) Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Investig New Drugs 17(4):361–373
Usmani SZ, Bona R, Li Z (2009) 17 AAG for HSP90 inhibition in cancer–from bench to bedside. Curr Mol Med 9(5):654–664
Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-Kaye M, Chen M, Steinberg SM, Muir CA, Yancey MA, Horneffer YR, Juwara L, Melillo G, Ivy SP, Merino M, Neckers L, Steeg PS, Conley BA, Giaccone G, Doroshow JH, Murgo AJ (2010) Phase I trial of 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), a heat shock protein inhibitor, administered twice weekly in patients with advanced malignancies. Eur J Cancer 46(2):340–347. doi:10.1016/j.ejca.2009.10.026
Shapiro GI, Kwak E, Dezube BJ, Yule M, Ayrton J, Lyons J, Mahadevan D (2015) First-in-human phase i dose escalation study of a second-generation non-ansamycin HSP90 Inhibitor, AT13387, in patients with advanced solid tumors. Clin Cancer Res 21(1):87–97. doi:10.1158/1078-0432.CCR-14-0979
Curry J, Angove H, Fazal L, Graham B, Harada I, Lyons J, Reule M, Smyth T, Thompson N (2009) Significance of long term pharmacodynamic actions of the HSP90 inhibitor AT13387. Proceedings of the AACR 100th Annual Meeting in Experimental and Molecular Therapeutics, Denver, CO: Abstract number 1856
Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC (1997) Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 89(15):1138–1147
Rajan A, Kelly RJ, Trepel JB, Kim YS, Alarcon SV, Kummar S, Gutierrez M, Crandon S, Zein WM, Jain L, Mannargudi B, Figg WD, Houk BE, Shnaidman M, Brega N, Giaccone G (2011) A phase I study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res 17(21):6831–6839. doi:10.1158/1078-0432.CCR-11-0821
Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM (2006) Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 5(10):2512–2521. doi:10.1158/1535-7163.MCT-06-0334
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18(1):64–76. doi:10.1158/1078-0432.CCR-11-1000
McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C (2006) Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res 66(22):10967–10975. doi:10.1158/0008-5472.CAN-06-1629
Smyth T, Paraiso KH, Hearn K, Rodriguez-Lopez AM, Munck JM, Haarberg HE, Sondak VK, Thompson NT, Azab M, Lyons JF, Smalley KS, Wallis NG (2014) Inhibition of HSP90 by AT13387 delays the emergence of resistance to BRAF inhibitors and overcomes resistance to dual BRAF and MEK inhibition in melanoma models. Mol Cancer Ther 13(12):2793–2804. doi:10.1158/1535-7163.MCT-14-0452
Acknowledgments
The authors thank Dr. Andrea Regier Voth, Leidos Biomedical Research, Inc., for medical writing support in the preparation of this manuscript, and Ms. Tracy Webb, Leidos Biomedical Research, Inc., for pharmacokinetic technical assistance. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and Core grant #CA16672. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. John Lyons is an employee of Astex Pharmaceuticals; no potential conflicts of interest were disclosed by the other authors.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Do, K., Speranza, G., Chang, LC. et al. Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Invest New Drugs 33, 921–930 (2015). https://doi.org/10.1007/s10637-015-0255-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0255-1