
Summary
The primary objective of this phase I study of LY2780301, a dual p70 S6 kinase and Akt inhibitor, was to determine the recommended phase II dose as a single agent in patients with advanced cancer. Secondary objectives included safety, pharmacokinetic, and pharmacodynamic analyses, and co-clinical analyses in Avatar models. Eligible patients received total daily doses of LY2780301 100–500 mg, given orally as a single dose or divided into 2 doses for 28-day cycles. Dose escalation followed 3 + 3 design. The primary pharmacodynamic endpoint was inhibition of S6 assessed by skin and tumor biopsy. Thirty-two patients were treated. Common toxicities possibly related to treatment included constipation (19 %), fatigue (13 %), nausea (9 %), and diarrhea (9 %). Grade 3/4 toxicities potentially related to treatment were anemia (n = 2), increased alanine aminotransferase/aspartate aminotransferase (ALT) (n = 1), and increased gamma-glutamyl transpeptidase (GGT) (n = 1). One patient experienced best overall response of prolonged stable disease for 6 cycles. Plasma exposures of LY2780301 exceeded predicted efficacious exposures, but were not dose proportional. Among patients receiving 500 mg daily >50 % exhibited reduced S6 in skin biopsies at Day 8 of treatment, but the effect was not maintained. Plasma concentrations of LY2780301 and/or its metabolites were not correlated with S6 expression in the epidermis. There was minimal antitumor activity against the model, CRC 019. Avatar models showed minimal pharmacodynamic effects consistent with the observed antitumor effects. This study suggests a dose of LY2780301 500 mg QD for future studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The phosphatidlyinositol-3-kinase (PI3K)/Protein Kinase B (PKB, Akt/mammalian target of the rapamycin (mTOR) signaling pathway is a key regulator of cell proliferation and survival [1], and has been described as a “master switch” for cell growth and proliferation [2]. The PI3K/Akt/mTOR signaling pathway is frequently mutated [3–5] and is constitutively activated in human tumors [6]. The Akt family of serine-threonine protein kinases includes three isoforms: Akt1, Akt2, and Akt3 [7–12]. Activation of the PI3K/Akt/mTOR pathway results in the activation of the mTOR complex 1 (mTORC1), and p70 S6 kinase (p70S6K) is a key effector of mTOR. Activation of p70S6K and subsequent phosphorylation of the S6 ribosomal protein (S6) up-regulates mRNA translation, which promotes sustained cell growth and proliferation. Blockage of PI3K/Akt/mTOR pathway activation has been shown to result in apoptosis or cell cycle arrest in several different models [13]. Therapeutic targeting of the PI3K/Akt/mTOR signaling pathway may provide a method of inhibiting protein synthesis similar to that of rapamycin and its analogues.
LY2780301 is a highly selective adenosine triphosphate (ATP)-competitive dual inhibitor of p70S6K and Akt. Preclinically, this small molecule exhibited antiproliferative activity in a broad range of cell lines using monolayer and colony formation assays (data on file, Eli Lilly and Company). LY2780301 effectively inhibited the growth of A2780 (ovarian), H460 (lung), PC3 (prostate), and HCT116 (colon) xenograft models. Pharmacodynamic relationships of LY2780301 with phospho-S6 (pS6) and other markers were dose-, exposure-, and time-dependent (data on file, Eli Lilly and Company).
This first-in-human phase I study of LY2780301 had a primary objective to determine the recommended phase II dose and schedule as an orally administered single agent in patients with advanced solid tumors. Secondary objectives included evaluation of safety, pharmacokinetics (PK), and pharmacodynamics (PD) of the compound and its metabolites. Parallel clinical and nonclinical investigations (co-clinical trials), which included Avatar mouse models of cancer, were conducted and included models with tumor tissues obtained from clinical study patients. Co-clinical studies provided relevant preclinical models to test the study agent for further mechanistic and preclinical studies. This approach has been used in clinical studies with novel anticancer agents and has resulted in a better understanding of mechanisms of action [14].
Patients and methods
Ethics statement
This study followed the guiding principles of the Declaration of Helsinki [15] and the Good Clinical Practice Guidelines of the International Conference on Harmonisation [16]. All patients provided written informed consent prior to study enrollment.
Patients
This multicenter Phase 1 study had the following patient eligibility criteria for enrollment: age ≥18 years; histologically confirmed solid tumors or Non-Hodgkin’s lymphoma (NHL) refractory to standard therapy; measurable or nonmeasurable disease defined by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) [17]; discontinued previous treatments for cancer and recovered from the acute effects of therapy for at least 28 days for myelosuppressive agents or 14 days for nonmyelosuppressive agents; a performance status of ≤1 on the Eastern Cooperative Oncology Group (ECOG) scale; and baseline laboratory tests to determine adequate hematopoietic, renal, and hepatic function [defined as absolute neutrophil count ≥1.5 × 109/L; platelets ≥100 × 109/L; hemoglobin ≥8 g/dL; serum creatinine ≤1.5×upper limits of normal (ULN) or calculated clearance >45 mL/min; bilirubin ≤1.5× ULN; and alanine transaminase (ALT) ≤2.5 x ULN (≤5× ULN was acceptable for patients with liver tumors)].
Patients were excluded for any of the following reasons: received treatment within 28 days of the initial dose of study drug with a drug that had not received regulatory approval for any indication; symptomatic central nervous system malignancy or metastasis (except for patients no longer receiving corticosteroids and/or anticonvulsants with asymptomatic and stable disease for at least 60 days); current acute or chronic leukemia; positive test results for human immunodeficiency virus, hepatitis A, B, or C; corrected QT interval (QTc) >470 msec on an electrocardiogram; treatment with a strong cytochrome P450 3A4 (CYP3A4) substrate with a narrow therapeutic range, or classification as a strong inhibitor or inducer; history of pituitary adenoma; or pregnancy or lactation.
Study design
This nonrandomized, open-label dose escalation phase I study of LY2780301 in patients with advanced solid tumors or NHL incorporated once-daily (QD) (Part A) and twice-daily (BID) (Part B) dose regimens as part of a 28-day dosing cycle. Dose escalation was to follow a 3 + 3 design until the criteria for maximum tolerated dose (MTD) were met. Part B initiation was based on patient toxicity and PK/PD data from Part A. Part A dosing began at 100 mg in a flat dosing scheme and escalated by 100 mg in each subsequent cohort, up to 500 mg. In Part B, dosing began at 150 mg BID and escalated by 50 mg for the second cohort (200 mg BID).
In Parts A and B, each cohort initially included 3 patients. At the end of the first treatment cycle, each patient was clinically evaluated for safety by the investigator before being allowed to receive the next treatment cycle. Eligible patients received 2 cycles of LY2780301, unless one or more criteria for discontinuation were met. Discontinuation criteria included: progressive disease; unacceptable toxicity; noncompliance of the patient; a dosing delay of more than 2 weeks due to an AE; a dose-limiting toxicity (DLT) leading to dose reduction with another DLT-equivalent toxicity occurring at the reduced dose in Cycle 2 or greater; and withdrawal by the patient, attending physician, or sponsor for any reason. Patients who, in the opinion of the investigator, demonstrated clinical benefit may have received treatment beyond 2 cycles.
Dose escalations were considered following an assessment of toxicity using the Common Terminology Criteria for Adverse Events Version 4(CTCAE) Version 4.02) [18]. Dose escalation decisions primarily considered any adverse events (AEs) possibly related to LY2780301, along with PK/PD data, when available, as a secondary consideration. Patients received at least two cycles of treatment unless one or more criteria for discontinuation were met. DLT was defined as an AE possibly related to LY2780301 occurring during cycle 1 following the CTCAE v4.02 criteria: Grade 4 thrombocytopenia or neutropenia >5 days’ duration; febrile neutropenia; ≥Grade 3 non-hematological toxicity except nausea/vomiting/diarrhea, skin rash that was responsive to medical treatment; transient (≤5 days) Grade 3 elevations of ALT/aspartate aminotransferase (AST) without evidence of other hepatic injury; transient Grade 3 hyperglycemia; and Grade 3 hypertriglyceridemia or hyperlipidemia without optimal treatment. If a single patient experienced DLT during Cycle 1 of LY2780301, three additional patients were enrolled at that dose level. If a DLT was observed in two or more patients at any dose level, escalation ceased and the previous dose was declared the MTD. An expansion cohort of up to 30 patients was planned once the recommended dose was reached.
Response analyses were based on RECIST Version 1.1 [17] for patients with solid tumors, or the Revised Response Criteria for Malignant Lymphoma or patients with non-Hodgkin’s lymphoma [19].
Drug supply
LY2780301 was provided by Eli Lilly & Company (Indianapolis, IN, USA) as capsules for oral administration containing 25 or 100 mg of active drug.
Pharmacokinetic studies
Pharmacokinetic analyses were performed on all patients who received at least one dose of study drug and contributed post-dose blood samples for bioanalysis according to the study protocol. Whole blood samples were collected pre-dose and at 0.5, 1, 2, 3, 5, and 8 h post-dose on days 1 and 8 of Cycle 1. Concentrations of LY2780301 parent drug and its two principal metabolites (desmethyl and didesmethyl) were measured by a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method [data on file, Eli Lilly and Company]. Pharmacokinetic parameters following single and multiple doses of LY2780301 included partial area under the plasma concentration-time curve (AUC), peak observed concentration (Cmax), time to Cmax (tmax), half-life (t½), apparent clearance (CL/F), and apparent steady-state volume of distribution (Vss/F).
Pharmacodynamic studies
Pharmacodynamic analyses were performed in skin biopsies collected on days 1, 8, and 22 of Cycle 1. The pS6 expression was investigated in two scoring schemes: 1) levels of pS6 expression in the entire epidermis, and 2) levels of pS6 expression in the epidermis minus the stratum granulosum (epidermis-SG).
Antitumor activity
Patients’ tumor measurements were assessed by one or more of the following radiologic tests: computerized tomography (CT) scan, magnetic resonance imaging (MRI), and/or chest x-ray. The extent of each patient’s disease was assessed using the following procedures: tumor measurement of palpable or visible lesions for patients with solid tumors (RECIST v1.1 guidelines), [17] or the Revised Response Criteria for Malignant Lymphoma [19] for patients with NHL.
Animal studies
The study protocol for the parallel co-clinical study in Avatar mouse models of cancer was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). The care and use of animals in this study complied with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Six-week old female athymic nude-Foxn1 nu mice (Harlan) were housed in individual high-efficiency particulate air (HEPA) ventilated cages (Sealsafe® Plus, Techniplast). Tumor fragments were subcutaneously implanted bilaterally on the right and left flank, and included colon cancer (three models: CRC 005, 019, 012), pancreatic cancer (three models: Panc 031, 198, 215), lung cancer (two models: Pulm 021, 024), and one melanoma model (Mel001) obtained from one of the study subjects. Animals were randomized into treatment and control groups when tumors reached ~200 mm3, at which point dosing was initiated (Day 1). LY2780301 was administered at a dose of 12.5 or 50 mg/kg daily Monday-Friday by oral gavage. Animals were checked daily for mobility, body weight, morbidity and other abnormal effects, and mortality. Tumor sizes were measured (in triplicate) twice weekly in two dimensions using an electronic caliper, and the tumor volume was expressed in mm3 using the formula TV = width 2 x length x 0.5. Percent tumor growth inhibition (%TGI) values were calculated for each treatment group (T) versus control (C) using initial (i) and final (f) tumor measurements by the equation %TGI = 1- [(Tf-Ti)/(Cf-Ci)]. TGI values were compared between treatment and control groups.
Western blot analysis of LY2780301
Total protein from xenograft samples was extracted with RIPA buffer (R0278, Sigma Aldrich, St. Louis, Missouri, USA) plus protease inhibitor cocktail tablet (11836170001, Roche Diagnostics, Indianapolis, Indiana, USA) and phosphatase inhibitor cocktail 2 and 3 (P5726, P0044 Sigma Aldrich, St. Louis, Missouri, USA). Analyses were conducted after SDS-PAGE electrophoresis and transferred to polyvinylidene difluoride Immobilon®-P membranes (Millipore®). Immunoblotting was performed according to the antibody manufacturers’ recommendations. The following antibodies were used: anti-S6 (2217), anti-pS6 (4857), anti-Akt (4691), anti-phospho-Akt (pAkt) (4060), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5174) (Cell Signaling Technology®, Danvers, Massachusetts, USA). Secondary antibodies included goat anti-rabbit-horseradish peroxidase (HRP) (P0448, Dako, Carpinteria, California, USA).
Results
Phase I study patient demographics and treatment
Thirty-two patients completed at least one cycle of LY2780301. The mean treatment durations were 2.9 cycles for both Parts A and B (range Part A: 1–9, Part B: 1–5). Most patients had numerous prior therapies; 72 % of patients had ≥3 previous lines of systemic treatment. Patient characteristics and the dose-escalation scheme are summarized in Tables 1 and 2, respectively.
Safety and tolerability
Twenty-five patients in Part A and seven patients in Part B received at least one dose of LY2780301. There were no DLTs reported in either Part A or Part B. Eight patients (25 %) experienced a serious adverse event (SAE) during treatment: 500 mg QD cohort (n = 3), 300 mg QD cohort (n = 2), 400 mg QD cohorts (n = 2), and 100 mg QD cohort (n = 1). The most common SAEs were dyspnea and respiratory failure, experienced by two patients each. One patient each experienced the following SAEs: gastrointestinal disorder, small intestine obstruction, enterocolitis infection, upper respiratory infection, acute kidney injury, renal colic, and surgical procedure. None of these SAEs were considered related to study drug.
For Parts A and B, 21 patients (66 %) reported at least one treatment-emergent adverse event (TEAE) possibly related to study drug (Table 2). There were four Grade 3/4 TEAEs possibly related to study drug: Grade 3 anemia (n = 1), increased ALT /AST (n = 1), increased GGT (n = 1), and Grade 4 anemia (n = 1). The most common TEAEs possibly related to study drug were constipation (19 %), fatigue (13 %), nausea (9 %), and diarrhea (9 %). Laboratory abnormalities possibly related to study drug included increased ALT/AST (3 %), hyperglycemia (6 %), anemia (6 %), and thrombocytopenia (3 %). No patients discontinued treatment due to an adverse event.
Pharmacokinetics
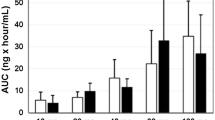
In Part A, LY2780301 exposures, as assessed by AUC from time zero to infinity following the first dose on day 1(AUC[0-∞]), increased with doses from 100 to 500 mg QD. A similar trend in AUC was observed during one dosing interval at steady state (AUC[τ,ss]) on Day 8, with the exception of the 200-mg QD cohort, in which the average AUC was slightly lower than the 100-mg QD cohort. Exposures across the QD dose range were not dose-proportional. Corresponding median tmax across treatment groups ranged from 3 to 6 h. PK results are summarized in Table 3.
Twice-daily dosing was evaluated for the 150-mg and 200-mg doses and compared with QD dosing at steady state (Day 8). Mean exposures in the former groups ranged from 36,700 to 39,000 ng · hr/mL (73,400-78,000 ng · hr/mL extrapolated over a 24-hour period). Although sample sizes were small, these results demonstrated BID dosing resulted in lower overall daily exposures compared to QD dosing. Therefore, additional BID dosing -regimens were not evaluated.
Using pooled data from Part A and Part B, a PK model determined the mean CL/F and Vss/F estimates ranged from 2.23 to 5.48 L/hr and from 43.4 to 207 L, respectively. Mean t1/2 ranged from 8.58 to 29.3 h, with the t1/2 generally increasing with increasing dose. Exposures at steady state were within the anticipated efficacious range of exposures based on preclinical data (data on file, Eli Lilly and Company).
Plasma concentrations of the desmethyl (LSN2804018) and didesmethyl (LSN2804027) metabolites of LY2780301, both of which are known to exhibit measurable, sub-potent activities relative to the parent molecule, were analyzed (data on file, Eli Lilly and Company). Median plasma concentrations of these metabolites varied among treatment groups, ranging from 5 to 29 % for the desmethyl metabolite, and from 0.07 to 0.89 % for the didesmethyl metabolite on a molar basis (Table 3).
Pharmacodynamics
The level of ribosomal pS6 inhibition was evaluated in pre-treatment and on-treatment skin biopsies via quantitative immunohistochemistry (IHC). Skin samples were taken from 15 patients at the highest administered doses. Eleven patients had samples available at baseline and also at Cycle 1 day 8, pre-dose and 5 h post-dose. Ten of the 11 patients were treated at the 500 mg QD dose, and one patient was treated at the 200 mg BID dose. In the 500 mg QD cohort, six of the ten patients achieved pS6 inhibition, which was in alignment with a pre-established relevant threshold. Seven of the ten patients had a decrease in pS6 expression levels post-LY2780301 when compared with baseline epidermis-SG measures. Six of these ten patients exhibited a decrease in pS6 levels using the entire epidermis measures (Fig. 1). However, the decrease in pS6 expression was transient and was not maintained in the 5-hour post-dose samples collected on Day 8 of cycle 1 (data on file, Eli Lilly and Company). Only two patients had pre- and post-dose pS6 levels measured on Day 22, and both of these patients exhibited a decrease in pS6 expression. (data on file, Eli Lilly and Company).

pS6 levels in skin biopsies. The graph represents change in pS6 levels in skin biopsies from the epidermis and epidermis minus the stratum granulosum (epidermis-SG) for patients in the 500-mg QD cohort at Cycle 1 day 8 postdose (C1D8_0hr) compared to baseline Each dot represents a single patient Epidermis SG = epidermis minus the stratum granulosum
Antitumor activity
There were 28 evaluable patients for disease response. Of these, 8 (29 %) patients exhibited stable disease. Five of the eight patients had a progression free survival (PFS) >120 days (mesothelioma n = 2, both 500 mg QD; breast cancer n = 2, 400 mg QD, 150 mg BID; colon cancer n = 1, 200 mg BID). There were no responders in this study. A molecular tumor profile is available for 10 of 32 patients, and is summarized in the Supplemental Table. One patient with colon cancer and PIK3CA E545K mutation had stable disease for 125 days.
Co-clinical analyses of LY2780301
Figure 2 shows the tumor growth inhibition curves of Avatar models treated with LY2780301 at the indicated doses. Overall tumor growth inhibition (TGI) and the genetic characteristics of the tumors tested are shown in Fig. 3. Overall, there was minimal antitumor activity against the model CRC 019, with a TGI of 68 and 64 % for the 12.5 and 50-mg/kg doses, respectively, on day 28 that was not maintained on day 50. Assessment of pathway inhibition in treated models showed minimal PD effects consistent with the observed antitumor effects (Fig. 3).

Tumor growth inhibition curves of LY2780301 against a series of Avatar mouse models of cancer Groups of xenograft tumors (with indicated tumor model) were treated at the dose and schedule indicated. Tumor sizes were monitored and tumor volumes determined as described in the Methods section CRC, colorectal cancer; Panc, pancreatic cancer; Pulm, pulmonary cancer; Mel, melanoma

Tumor xenograft characteristics and expression of pS6 and pAkt. TOP Expression of (p)S6 and (p)Akt in xenograft samples from control (C) mice and mice treated for 29 days with LY2780301 (T), analyzed by western blot BOTTOM Genetic characteristics of the patient-derived tumor xenografts from (Pulm021) (Panc198) and (CRC012) Pulm, pulmonary cancer; Panc, pancreatic cancer; CRC, colorectal cancer
Discussion
The primary objective of this phase I study was to recommend a phase II dose and schedule for LY2780301. However, because there were no DLTs, the MTD was not identified. Based on PK, PD, and clinical results, the dose of 500 mg QD was recommended for future studies . Signs of clinical benefit were observed in a few patients at different dose levels with different pretreated and progressing tumor types, including mesothelioma and breast cancer.
Mean PK exposures of LY2780301 at all dose levels exceeded the a priori established threshold of efficacious exposure based on preclinical data (>25,000 ng · hr/mL) and PK modeling. This might explain the high variability of exposure to LY2780301 and the lack of apparent dose proportionality. It was hypothesized that variability in exposures was related to variability of dissolved LY2780301 in the gastric compartment, a phenomenon that is pH-dependent. The terminal slopes for the plasma concentration-time curves across dose ranges were approximately parallel. The absence of dose proportionality was likely due to limited absorption of the higher dose because of low solubility. The sizable inter-individual variability within cohorts and sparse sampling (8 h or- 24-hours post-dose) precluded formal assessment of dose proportionality and limited characterization of the terminal elimination phase. The apparent trend for t1/2 likely was caused by the dose-dependent increase in Vss/F. Median accumulation, as calculated by the ratio of AUC from time zero to 8 h [AUC(0–8)] on day 8 relative to day 1, ranged from 1.37 to 3.32.
There was evidence to suggest that LY2780301, at the recommended dose of 500 mg QD, affected IHC expression of pS6 in post-LY2780301 skin biopsies. Unfortunately, pS6 inhibition data were not obtained throughout the different dose regimens, limiting the possibility of describing an exposure-response relationship. At the highest dose levels in both schemes, PK/PD plots showed no visible correlation between inhibition of pS6 levels in the epidermis and exposure to LY2780301 or its desmethyl and didesmethyl metabolites.
Conducting parallel clinical and co-clinical investigations has recently been proposed as a strategy to optimize drug development [20–22]. In previous studies, co-clinical models enabled recapitulation of the heterogeneity of human cancer and were predictive of clinical outcome [14, 23, 24]. In this study, at the dose and schedules tested, the LY2780301 was not effective against any selected models (Fig. 2). This suggests that additional doses, schedules, or combinations would be needed for therapeutic efficacy. Of particular interest was model Mel001 developed from a patient with BRAF wild-type malignant melanoma (Fig. 3). The patient developed disease progression after one cycle of treatment, which was consistent with the lack of activity of LY2780301 in the patient’s corresponding Avatar model. Patients in this trial were not selected based on their tumor molecular profile; no valid hypothesis was available at the time of study initiation to select patients based on tumor molecular profiling. The analysis of limited tumor profile data did not show a clear link between PI3K mutations and duration of stable disease.
There are several known therapeutic inhibitors of the PI3K/Akt/mTOR signaling pathway. Rapamycin and its analogues temsirolimus/CCI-779 (Torisel®, Wyeth Pharmaceuticals, Madison, NJ, USA) [25–27], everolimus/RAD 001 (Afinitor®, Novartis Pharma, Basel, Switzerland) [28], and ridaforolimus/A23573 (Ariad Pharma, Cambridge, MA, USA) [29] allosterically inhibit mTORC1. Following the approval of everolimus and temsirolimus for treating breast and renal cancer, other similar compounds were developed, including: ATP-competitive, dual inhibitors of class I PI3K and mTORC1/2; ‘pan-PI3K’ inhibitors that inhibit all four isoforms of class I PI3K (α, β, δ, γ); isoform-specific inhibitors of the various PI3K isoforms; allosteric and catalytic inhibitors of Akt; and ATP-competitive inhibitors of mTOR only. While these compounds block the same signaling pathway, they have different levels of antitumor activity and varying levels of toxicity depending on the tumor’s genetic context.
Similarly, ATP-competitive dual inhibitors of class I PI3K and mTORC1/2 may have a broader activity profile, but toxicity may preclude these compounds from attaining sufficient therapeutic doses. On the opposite extreme of selectivity, isoform-specific inhibitors may be only effective in specific contexts. INK-1402, a selective p110 α inhibitor, was more effective in PI3K catalytic subunit α (PI3KCA) mutated cell lines compared with mutated or absent PTEN. BYL719 (Novartis) and GDC-0032 (Genentech) reported partial responses exclusively in patients with PI3K3CA mutant tumors [30, 31]. Preclinical work showed that Akt inhibitors provide an interesting strategy for tumors with either PIK3CA or PTEN alterations [32–37]. Combined treatment with a PI3K-alpha inhibitor and an mTOR inhibitor was found to be synergistic in PIK3CA mutant tumors [38]. This provides a rationale for future trials in combination with other anticancer agents and molecular selection of patients.
References
Takeuchi CS, Kim BG, Blazey CM MAS, Johnson HWB, Anand NK, Arcalas A, Baik TG, Buhr CA, Cannoy J, Epshteyn S, Joshi A, Lara K, Lee MS, Wang L, Leahy JW, Nuss JM, Aay N, Aoyama R, Foster P, Lee J, Lehoux I, Munagala N, Plonowski A, Rajan S, Woolfrey J, Yamaguchi K, Lamb P, Miller N (2013) Discovery of a novel class of highly potent, selective, ATP-competitive, and orally bioavailable inhibitors of the mammalian target of rapamycin (mTOR). J Med Chem 56:2218–2234
Zhou H, Luo Y, Huang S (2010) Updates of mTOR inhibitors. Anticancer Agents Med Chem 10:571–581
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB (2005) Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 4:988–1004
Rosen N, She QB (2006) AKT and cancer–is it all mTOR? Cancer Cell 10:254–256
Shaw RJ, Cantley LC (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441:424–430
Bartholomeusz C, Gonzales-Angulo AM (2012) Targeting the Pi3K signaling pathway in cancer therapy. Expert Opin Ther Targets 16:121–130
Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N (2001) Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 15:2203–2208
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ (2001) Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276:38349–38352
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ (2005) Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol 25:1869–1878
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest 112:197–208
Gonzalez E, McGraw TE (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8:2502–2508
Tschopp O, Yang ZZ, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, Michaelis T, Frahm J, Hemmings BA (2005) Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 132:2943–2954
Vivanco I, Sawyers C (2002) The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev Cancer 2:489–501
Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, Zhang H, Soon-Shiong P, Shi T, Rajeshkumar NV, Maitra A, Hidalgo M (2011) Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 29:4548–4554
World Medical Assembly. Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. Available at: www.wma.net/en/30publications/10policies/b3/. Accessed 9 February 2015
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. International Conference on Harmonization (ICH) Guideline for Good Clinical Practices- E6(R1) (1996). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. Accessed 9 February 2015
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
U.S. Department of Health and Human Services National Institutes of Health National Cancer Institute (2009) Common Terminology Criteria for Adverse Events (CTCAE) Version 4.02 NIH Publication No. 03–5410. http://evs.nci.nih.gov/ftp1/CTCAE/Archive/CTCAE_4.02_2009-09-15_QuickReference_8.5x11pdf 15. Accessed 9 February 2015
Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, Rosen ST, Stroobants S, Lister TA, Hoppe RT, Dreyling M, Tobinai K, Vose JM, Connors JM, Federico M, Diehl V, International Harmonization Project on Lymphoma (2007) Revised response criteria for malignant lymphoma. J Clin Oncol 25:579–586
Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, Gao P, Woo MS, Xu C, Yanagita M, Altabef A, Wang S, Lee C, Nakada Y, Peña CG, Sun Y, Franchetti Y, Yao C, Saur A, Cameron MD, Nishino M, Hayes DN, Wilkerson MD, Roberts PJ, Lee CB, Bardeesy N, Butaney M, Chirieac LR, Costa DB, Jackman D, Sharpless NE, Castrillon DH, Demetri GD, Jänne PA, Pandolfi PP, Cantley LC, Kung AL, Engelman JA, Wong KK (2012) A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483:613–617
Garrido-Laguna I, Tan AC, Uson M, Angenendt M, Ma WW, Villaroel MC, Zhao M, Rajeshkumar NV, Jimeno A, Donehower R, Iacobuzio-Donahue C, Barrett M, Rudek MA, Rubio-Viqueira B, Laheru D, Hidalgo M (2010) Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer 103:649–655
Nardella C, Lunardi A, Patnaik A, Cantley LC, Pandolfi PP (2011) The APL paradigm and the “co-clinical trial” project. Cancer Discov 1:108–116
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L (2011) A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1:508–523
Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, Tanaka K, Singh S, Salimi-Moosavi H, Bouraoud N, Amador ML, Altiok S, Kulesza P, Yeo C, Messersmith W, Eshleman J, Hruban RH, Maitra A, Hidalgo M (2006) An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 12:4652–4661
Del Bufalo D, Ciuffreda L, Trisciuoglio D, Desideri M, Cognetti F, Zupi G, Milella M (2006) Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res 66:5549–5554
Dudkin L, Dilling MB, Cheshire PJ, Harwood FC, Hollingshead M, Arbuck SG, Travis R, Sausville EA, Houghton PJ (2001) Biochemical correlates of mTOR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin Cancer Res 7:1758–1764
Geoerger B, Kerr K, Tang CB, Fung KM, Powell B, Sutton LN, Phillips PC, Janss AJ (2001) Antitumor activity of the rapamycin analog CCI-779 in human primitive neuroectodermal tumor/medulloblastoma models as single agent and in combination chemotherapy. Cancer Res 61:1527–1532
Afinitor®: East Hanover, NJ. Novartis Pharmaceuticals Corp. 2012
Squillace RM, Miller D, Cookson M, Wardwell SD, Moran L, Clapham D, Wang F, Clackson T, Rivera VM (2011) Antitumor activity of ridaforolimus and potential cell-cycle determinants of sensitivity in sarcoma and endometrial cancer models. Mol Cancer Ther 10:1959–1968
Juric D, Rodon J, Gonzalez-Angulo A, Burris HA, Bendell J, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J (2012) BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res 72 (Suppl 1, abstract CT-01)
Ndubaku CO, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, Bull R, Do S, Dotson J, Dudley D, Edgar KA, Friedman LS, Goldsmith R, Heald RA, Kolesnikov A, Lee L, Lewis C, Nannini M, Nonomiya J, Pang J, Price S, Prior WW, Salphati L, Sideris S, Wallin JJ, Wang L, Wei B, Sampath D, Olivero AG (2013) Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J Med Chem 56:4597–4610
Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, Ricketts SA, Cross D, Cosulich S, Chresta CC, Page K, Yates J, Lane C, Watson R, Luke R, Ogilvie D, Pass M (2012) Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 11:873–887
Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, Lee LB, Risom T, Gross S, Liederer BM, Koeppen H, Skelton NJ, Wallin JJ, Belvin M, Punnoose E, Friedman LS, Lin K (2013) Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res 19:1760–1772
Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, Miller TW, Arteaga CL, Mills GB, Gonzalez-Angulo AM, Meric-Bernstam F (2012) Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res 18:5816–28
She QB, Chandarlapaty S, Ye Q, Lobo J, Haskell KM, Leander KR, DeFeo-Jones D, Huber HE, Rosen N (2008) Breast tumor cells with PI3K mutation or HER2 amplification are selectively addicted to Akt signaling. PLoS One 3, e3065
Yap TA, Walton MI, Hunter LJ, Valenti M, de Haven BA, Eve PD, Ruddle R, Heaton SP, Henley A, Pickard L, Vijayaraghavan G, Caldwell JJ, Thompson NT, Aherne W, Raynaud FI, Eccles SA, Workman P, Collins I, Garrett MD (2011) Preclinical pharmacology, antitumor activity, and development of pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930. Mol Cancer Ther 10:360–371
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29:4688–4695
Elkabets M1, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, Serra V, Rodrik-Outmezguine V, Hazra S, Singh S, Kim P, Quadt C, Liu M, Huang A, Rosen N, Engelman JA, Scaltriti M, Baselga J (2013) mTORC1 Inhibition Is Required for Sensitivity to PI3K p110alpha Inhibitors in PIK3CA-Mutant Breast Cancer. Sci Transl Med 5: 196ra99
Acknowledgments
The study was sponsored by Eli Lilly and Company. The authors acknowledge Suzanne R.L. Young, Ph.D., and Jennifer M. Harris, Pharm.D., employees of Eli Lilly and Company, for writing assistance. Antonio Calles is a “Rio Hortega” fellowship grant recipient from the Instituto de Salud Carlos III (CM09/00283).
Conflicts of interest
A.A., J.R., A.C., I.B., P.P.L.C., and J.M. have no conflicts of interest to report. W.B. is an employee and shareholder of Onyx Pharmaceuticals, a subsidiary of Amgen, and is a shareholder of Eli Lilly and Company. M.H. received research support and an honorarium from Eli Lilly and Company. E.C. received clinical research support and an honorarium (Advisory Board) from Eli Lilly and Company. P.W., B.A.M., U.O., and K.A.B... are employees and shareholders of Eli Lilly and Company.
Previous disclosure
Data from this study, NCT01115751, were communicated as an oral presentation at The American Society of Clinical Oncology (ASCO) 2012 [J Clin Oncol 30, 2012 (suppl; abstr 3005).
Author information
Authors and Affiliations
Corresponding author
Additional information
At the time of this study, Dr. Miller and Mr. Bumgardner were employed by Eli Lilly and Company. Dr. Miller is currently employed by Everest Clinical Research, Little Falls, NJ, USA. Mr. Bumgardner is currently employed by Onyx Pharmaceuticals, a subsidiary of Amgen, South San Francisco, CA, USA.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 54 kb)
Rights and permissions
About this article

Cite this article
Azaro, A., Rodon, J., Calles, A. et al. A first-in-human phase I trial of LY2780301, a dual p70 S6 kinase and Akt Inhibitor, in patients with advanced or metastatic cancer. Invest New Drugs 33, 710–719 (2015). https://doi.org/10.1007/s10637-015-0241-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0241-7