
Summary
Purpose ABT‐806, a humanized recombinant monoclonal antibody, binds a unique epidermal growth factor receptor (EGFR) epitope exposed in the EGFRde2‐7 (EGFRvIII) deletion mutant and other EGFR proteins in the activated state. This phase I study evaluated the safety, pharmacokinetics, and recommended phase two dose (RP2D) of ABT-806 in patients with solid tumors that commonly overexpress activated EGFR or EGFRvlll. Methods Patients with advanced solid tumors, including glioblastoma, were eligible. Following a dose escalation phase, expanded safety cohorts of patients with solid tumors or EGFR-amplified glioblastoma were enrolled. Adverse events (AEs) were graded by National Cancer Institute Common Terminology Criteria for Adverse Events v4.0; tumor response was assessed by Response Evaluation Criteria in Solid Tumors v1.1. EGFR protein expression was quantified by immunohistochemistry. Results 49 patients were treated. Frequent AEs (≥10 %) possibly/probably related to ABT-806 were fatigue (18 %), nausea (16 %), dermatitis acneiform (12 %), and vomiting (10 %). Only one dose-limiting toxicity (grade three morbilliform rash) occurred. The RP2D was the pre-specified highest dose (24 mg/kg). Systemic exposures were dose proportional between 2 and 24 mg/kg. Median time to progression was 55 days (95 % confidence interval, 53–57) in all patients and 43 days (22–57) for glioblastoma patients. No objective responses occurred; however, two patients had prolonged stable disease. An EGFR-amplified penile cancer patient has stable disease lasting over 2.5 years. Conclusions ABT-806 has unique pharmacokinetic and safety profiles. Toxicities were infrequent and typically low grade at the RP2D. Linear ABT-806 pharmacokinetics suggest lack of significant binding to wild-type EGFR in normal tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tyrosine kinase signaling is responsible for triggering growth and proliferation during normal physiologic development. Many human cancers exploit these signaling pathways through activation of growth factors and receptors in the tyrosine kinase family [1]. The epidermal growth factor receptor (EGFR) is a member of the transforming growth factor receptors, known to be dysregulated in a variety of epithelial cancers [2]. When activated, EGFR signals through mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI-3 kinase) pathways, leading to proliferation, survival, and metastasis [3].
EGFR overexpression has been linked to breast, brain, head and neck, colon, penile, squamous cell lung, and pancreatic cancers, among others [4, 5]. Glioblastoma (GBM), the most common malignant brain tumor in adults, is highly aggressive. EGFR is overexpressed in approximately 50 % of primary GBM tumors [6]. Genetic alterations conferring constitutive activity to the receptor are also enriched in the subset of GBM tumors harboring EGFR amplification [6]. Although the specific type of mutation is variable, the most common form, EGFRvIII, lacks the ligand-binding domain from exons 2–7, conferring unregulated kinase activity [6, 7]. Patients with GBM have a poor prognosis, making EGFR a promising target in this and other populations with EGFR overexpression.
Two classes of anti-EGFR drugs have been approved for a number of cancer types, including monocolonal antibodies (mAbs) that block EGFR ligand binding (eg, cetuximab, panitumumab) and EGFR-directed tyrosine kinase inhibitors (eg, erlotinib, gefitinib, and afatinib). Although these therapies have demonstrated clinical efficacy, including prolongation of progression-free survival (PFS) and/or overall survival (OS), nearly all patients eventually develop resistance [8, 9]. In addition, a high frequency of dermatologic toxicities, most commonly a skin rash resembling acne, can impact patient quality of life and affect patient compliance [10].
Currently available antibody-based therapies target wild-type EGFR by binding to an exposed domain on the receptor to prevent dimerization and activation. Studies with cetuximab demonstrate that antibody binding induces EGFR cellular internalization and degradation, causing a decrease in proliferative signaling through Erk-1 and −2, as well as increased pro-apoptotic signaling through activation of Bax and downregulation of Bcl-2 [11, 12]. Although selective for EGFR, this approach targets all expressed receptors at the cell surface irrespective of the activation state. In addition, because EGFR is highly expressed in the skin, many anti-EGFR antibody therapies require saturating doses to reach steady-state serum levels and achieve a therapeutic effect [13].
ABT-806 is a novel, humanized recombinant immunoglobulin (Ig) G1ĸ mAb that binds to a unique EGFR epitope exposed in the constitutively active EGFRvIII deletion mutant and EGFR proteins in the activated state [14, 15]. The activity of the receptor is inhibited by binding of ABT-806 to this epitope; however, this epitope is largely inaccessible when wild-type EGFR is expressed at normal physiologic levels [14]. This unique mechanism of inhibition may limit off-target activity resulting from wild-type receptor downregulation and decrease the frequency of common toxicities seen with non-selective EGFR inhibiting agents.
ABT-806 has demonstrated antitumor activity in a range of different preclinical cancer models, including mouse xenografts derived from EGFR-amplified glioma cells, orthotopic models of GBM, and de novo non–small cell lung cancer (NSCLC) models [15–17]. A phase one study using a labeled chimeric version of ABT-806 has also shown specific targeting of tumor tissue in both solid tumors and gliomas, in addition to demonstrating linear pharmacokinetics (PK) and mild toxicities [18]. Consequently, a study to evaluate ABT-806 in patients with advanced solid tumors, including GBM, was initiated.
Methods
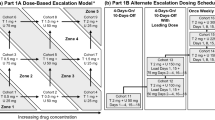
This was a phase 1, open-label study evaluating the safety, PK, and preliminary efficacy of ABT-806 in patients with advanced solid tumors that have historically been associated with either overexpression of activated EGFR or the presence of EGFRvIII. The first portion of the study evaluated the safety and PK profile of ABT-806 through escalating doses (Fig. 1). The second portion of the study included an expanded safety cohort that evaluated ABT-806 at the recommended phase two dose (RP2D) in patients with advanced solid tumors likely to overexpress activated EGFR or EGFRvlll, including patients with EGFR-amplified GBM (Fig. 1). Intravenous (IV) dosing began at 2 mg/kg every other week (eow). Based on safety and PK results, IV dosing continued to escalate to 6, 12, 18 and 24 mg/kg eow.

Study design. EGFR = epidermal growth factor receptor; GBM = glioblastoma; eow = every other week; RPn = recommended phase two dose
Eligible patients were ≥18 years of age with a solid tumor type known to either overexpress activated EGFR or to express EGFRvlll. Patients had measurable lesions, Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2, adequate organ function, and tumor tissue available for pharmacodynamic analysis. Patients with GBM were excluded from the dose escalation portion of the study, but were enrolled in the expanded safety portion. All patients with GBM were required to have demonstrated EGFR amplification on archival tumor tissue. Patients were not eligible if they were deemed to be at high risk for toxicities, had uncontrolled central nervous system metastases, or had received prior anticancer therapy within 21 days or prior EGFR-directed monoclonal antibody therapy within 4 weeks of study start. All patients provided written informed consent. This study was approved by local institutional review boards and was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. The ClinicalTrials.gov indentifier is NCT01255657.
Dose escalation was conducted using a 3 + 3 design. Escalation proceeded if the first three evaluable patients in a cohort completed 4 weeks of dosing without experiencing a dose-limiting toxicity (DLT). If one patient experienced a DLT, then three more patients were enrolled into the cohort and were evaluated. Escalation could continue, provided no additional patients experienced a DLT at that dose level. The maximum tolerated dose (MTD) was defined as the highest dose level at which fewer than two of six patients (<33 %) experienced a DLT. If the MTD was not reached, the RP2D was to be determined from overall safety and PK results. The highest dosing cohort, 24 mg/kg, was chosen because the projected human exposure was comparable to exposures in mice at the maximally efficacious dose. In addition, the highest dose tested was also the highest possible dose formulation; therefore, the RP2D could not exceed 24 mg/kg.
Plasma samples were collected at weeks one and seven for determination of ABT-806 exposure (Cmax), terminal elimination half-life (t1/2), and area under the serum concentration–time curve (AUC) from predose to 14 days (AUC14), using noncompartmental methods. An analysis of variance (ANOVA) was performed on PK variables to assess dose proportionality and linear kinetics.
Radiographic assessments for disease status were performed at baseline (within 21 days of study start) and repeated approximately every 8 weeks until the final visit. Tumor response was measured by the Response Evaluation Criteria in Solid Tumors (RECIST, 1.1). Time to progression (TTP), defined as the number of days from the date of the first dose to the date of disease progression, was analyzed using Kaplan-Meier methodology. Adverse events (AEs) were monitored and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, version 4.0).
Archival tumor samples were assessed for EGFR protein expression by immunohistochemistry (H-score) using the pharmDx EGFR kit (DAKO, Carpinteria, CA). A membrane H-score was calculated according to published methods [19]. Membrane staining was assessed visually and was scored by intensity. H-score was calculated by the addition of the percentage of weak staining cells (x 1), moderate staining cells (x 2), and strong staining cells (x 3). The H-score and days on study were compared to determine if an association existed between EGFR expression and the length of time a patient remained on study.
Results
Baseline characteristics
A total of 49 patients were enrolled at three sites in the United States, including 26 patients in the dose-escalation portion of the study and 23 patients in the expanded safety cohort. In the expanded safety cohort, 12 patients had advanced solid tumors and 11 had GBM. The median age of patients was 61 years (range, 39–81 years) and the majority of patients had an ECOG performance score of 1 (Table 1). The most common tumor types were GBM (22 %), NSCLC (22 %), colon (14 %), and head and neck (14 %). The most common reason for discontinuation was progressive disease (67 % of patients had radiographic progression and 22 % of patients had clinical progression).
Dose-limiting toxicities and determination of RP2D
Twenty-six patients were enrolled in five dose escalation cohorts: 2, 6, 12, 18, and 24 mg/kg. No DLTs were observed at the 2, 6, 12, and 18 mg/kg dose levels. One patient treated with 24 mg/kg ABT-806 experienced a DLT of grade three morbilliform rash that the investigator deemed as possibly related to the study drug. This rash was not acneiform and resolved with the administration of loratidine. No other DLTs occurred; therefore, the MTD was not reached. Given the favorable PK and safety profile, the RP2D was determined to be the pre-specified highest dose, 24 mg/kg eow.
Safety
Common treatment-emergent AEs and grade 3/4 AEs reported during the dose escalation phase and each arm of the expanded safety portion of the study were reported in Table 2. The most frequently reported AEs (in ≥10 % of patients) that were considered possibly or probably related to ABT-806 treatment were fatigue (18 %), nausea (16 %), dermatitis acneiform (12 %), and vomiting (10 %). One event each of atrial fibrillation, rash (morbilliform), and hypophosphatemia were grade three or four in severity and were deemed possibly or probably related to treatment with ABT-806. In total, six patients (12 %) developed dermatitis; all of these acneiform rashes were grade one.
Ten patients (39 %) during the dose escalation and 8 (35 %) in the expanded safety portions of the study experienced serious AEs. Two of these events occurred in patients in the GBM cohort and both were related to disease progression. Two serious events, grade three atrial fibrillation and grade two constipation, occurred during the dose escalation period and comprised the only serious events deemed at least possibly related to ABT-806 treatment. A total of 14 patients died during the course of the study, all due to disease progression. All treatment-emergent AEs that resulted in death were not considered to be related to study drug.
Pharmacokinetics
ABT-806 exhibited biphasic disposition after IV administration (Fig. 2). Systemic exposure (Cmax and AUC14) was approximately dose proportional over the studied dose range of 2 mg/kg to 24 mg/kg (Fig. 2 and Table 3). The harmonic mean for the terminal elimination phase half-life was 9.0 days following dosing on week 1 day 1. The PK of ABT-806 in the expanded safety cohort A (patients with advanced solid tumors) and cohort B (patients with GBM) were comparable to those of patients with solid tumors in the 24-mg/kg dose escalation cohort (data not shown).

Pharmacokinetics of ABT-806: a mean ± SD for ABT-806 AUC∞ versus ABT-806 dose on week 1, day 1 and b mean ± SD for ABT-806 AUC14 versus ABT-806 dose on week 7, day 1. AUC = area under the concentration–time curve; SD = standard deviation
Exploratory efficacy and biomarkers
The median time to progression was 55 days (95 % CI, 53–57 days) in all patients and 43 days (95 % CI, 22–57 days) for patients with GBM. No patients achieved a partial or a complete response; however, one patient with lung adenocarcinoma had stable disease for more than 6 months and one patient with penile cancer continues to have stable disease after more than 2.5 years on therapy. Data were available for 35 patients (69 %) whose EGFR expression was determined by computing average H-scores. H-scores were then plotted against each patient’s number of days on study (Fig. 3). No clear association existed between EGFR protein expression and duration of treatment or time on study. However, both patients with prolonged stable disease had a somatic EGFR genetic aberration. The patient with lung adenocarcinoma had a tumor harboring an EGFR exon 20 insertion mutation and the patient with penile cancer had a tumor harboring EGFR amplification.

Membrane H-score versus number of days on study
Discussion
This study was a first-in-human study evaluating the safety and tolerability of ABT-806. ABT-806 was well tolerated and dose escalation was able to continue to the pre-specified highest dose, leaving the highest dose tested, 24 mg/kg, as the RP2D. At this dose, toxicities were mainly grade one and two, including the AEs in patients with GBM.
Preclinical data suggest that the mechanism of action of ABT-806 differs from other Food and Drug Administration–approved EGFR antibodies. ABT-806 selectively binds to the activated form of EGFR and the EGFRvIII deletion mutant. These preclinical findings suggest that ABT-806 would not significantly bind to inactive EGFR proteins present in the skin [20], which has been confirmed by H-scores. Consistent with this prediction, our data show that ABT-806 had a low level of cutaneous toxicity. Four studies with cetuximab have noted a high rate of acneiform rashes (76 %–88 %) [21], with as many as 17 % of these rashes graded as severe. In this trial, however, low-grade dermatitis acneiform occurred in six patients (12 %). The only serious dermatologic AE was one grade three morbilliform rash, which had a very different morphology than the typical acneiform rash associated with EGFR inhibitors. Notably, the rash resolved with antihistamine therapy.
The favorable safety profile coincided with a linear PK of ABT-806 throughout the dose range tested, indicating the absence of extensive EGFR binding in normal tissues. This finding is again consistent with preclinical data that suggest that ABT-806 does not bind to non-activated EGFR proteins present in the skin. This is in contrast to published studies using cetuximab or panitumumab, which demonstrate nonlinear PK [22–24]. The high clearance rate of these antibodies at non-saturating doses has been attributed to high levels of tissue deposition, as wild-type EGFR expressed in the skin sequesters antibody from the systemic circulation. The absence of a high incidence of dermatologic adverse events, as well as the linearity of the PK of ABT-806, suggest that wild-type EGFR binding in normal tissues is minimal.
ABT-806 did not show significant activity among patients with heavily pretreated GBM with EGFR amplification. This arm of the expansion cohort was selected because approximately 50 % of patients with GBM with EGFR amplification have an EGFRvIII deletion [25]. Recent work has also shown that GBM tumors are molecularly heterogeneous, with mutually exclusive subpopulations harboring amplifications of EGFR and other tyrosine kinases, such as platelet-derived growth factor receptor (PDGFR), existing in individual tumors [26]. Although preclinical and clinical data have demonstrated that ABT-806 binds to GBM tumors, it is possible that inhibition of EGFR signaling alone is not an effective strategy for triggering cell death in this tumor type [17, 18].
Although no objective responses were observed, there were at least two patients with solid tumors with prolonged stable disease. Both of these patients had tumors with somatic EGFR genetic aberration, including a patient with lung adenocarcinoma containing EGFR exon 20 insertion mutation (EGFR D770_N771insGL) who remained on trial for more than 6 months, and a patient with EGFR-amplified penile cancer, who remains on trial after 2.5 years. The prolonged stable disease seen in the patient with NSCLC is noteworthy. Patients with lung adenocarcinoma with EGFR exon 20 insertion mutations typically do not respond to erlotinib or gefitinib and new molecularly targeted agents are needed for this population [27, 28].
One possible explanation why more activity was not seen with ABT-806 was that the population studied was heavily pretreated. Development of resistance to EGFR therapy is common and, in this trial, 55 % of the patients without GBM tumors were previously treated with EGFR inhibitors. Notably, the patient with EGFR-amplified penile cancer had never been treated with an EGFR inhibitor. Squamous carcinoma of the penis is a rare disease with limited therapeutic options. Several papers have reported EGFR overexpression and case series have been published describing the efficacy of EGFR inhibitors in this population [29–33]. Clinical trials studying EGFR inhibitors are needed to clearly establish the efficacy of this class of drugs in penile cancers.
Although preliminary antitumor activity was an exploratory objective, no correlation existed between tumor EGFR expression and the number of days a patient remained on study. This subset analysis was limited by the phase one design and overall low patient numbers. Tumor tissue was not tracked for changes in EGFR expression; therefore, it is not known whether ABT-806 induced a significant change in the expression or activity of the receptor. Further study is required to better define antibody targeting and its effect on receptor activity in tumor tissue. Taking into account the promising preclinical data and the PK and safety profile demonstrated in this study, it appears that ABT-806 may effectively target activated or mutant EGFR in tumor tissue without significant binding to normal tissue. This holds the promise for the use of ABT-806 as a payload delivery antibody, with such payloads as radiolabeling for diagnostic and predictive purposes and toxins to increase potency. Both such drug conjugates are currently being evaluated in ongoing clinical trials. In addition, the favorable toxicity profile of ABT-806 makes it an ideal candidate to be combined with other targeted agents in multi-drug regimens.
References
Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353:172–187
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358:1160–1174
Wong RW (2003) Transgenic and knock-out mice for deciphering the roles of EGFR ligands. Cell Mol Life Sci 60:113–118
Bonner JA, Raisch KP, Trummell HQ, Robert F, Meredith RF, Spencer SA, Buchsbaum DJ, Saleh MN, Stackhouse MA, LoBuglio AF, Peters GE, Carroll WR, Waksal HW (2000) Enhanced apoptosis with combination C225/radiation treatment serves as the impetus for clinical investigation in head and neck cancers. J Clin Oncol 18:47S–53S
Baselga J, Mendelsohn J (1994) The epidermal growth factor receptor as a target for therapy in breast carcinoma. Breast Cancer Res Treat 29:127–138
Frederick L, Wang XY, Eley G, James CD (2000) Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 60:1383–1387
Sugawa N, Ekstrand AJ, James CD, Collins VP (1990) Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A 87:8602–8606
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 25:1658–1664
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357:2040–2048
Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME (2007) Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology 72:152–159
Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J (2001) Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res 61:6500–6510
Huang SM, Bock JM, Harari PM (1999) Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res 59:1935–1940
Thomas SM, Grandis JR (2004) Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat Rev 30:255–268
Gan HK, Burgess AW, Clayton AH, Scott AM (2012) Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res 72:2924–2930
Luwor RB, Johns TG, Murone C, Huang HJ, Cavenee WK, Ritter G, Old LJ, Burgess AW, Scott AM (2001) Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res 61:5355–5361
Li D, Ji H, Zaghlul S, McNamara K, Liang MC, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Scott AM, Jungbluth AA, Cavenee WK, Old LJ, Demetri GD, Wong KK (2007) Therapeutic anti-EGFR antibody 806 generates responses in murine de novo EGFR mutant-dependent lung carcinomas. J Clin Invest 117:346–352
Mishima K, Johns TG, Luwor RB, Scott AM, Stockert E, Jungbluth AA, Ji XD, Suvarna P, Voland JR, Old LJ, Huang HJ, Cavenee WK (2001) Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res 61:5349–5354
Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, Papenfuss AT, Poon AM, Hopkins W, Smyth FE, MacGregor D, Cher LM, Jungbluth AA, Ritter G, Brechbiel MW, Murphy R, Burgess AW, Hoffman EW, Johns TG, Old LJ (2007) A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A 104:4071–4076
Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, de Marinis F, Eberhardt WE, Paz-Ares L, Storkel S, Schumacher KM, von Heydebreck A, Celik I, O’Byrne KJ (2012) EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol 13:33–42
Panousis C, Rayzman VM, Johns TG, Renner C, Liu Z, Cartwright G, Lee FT, Wang D, Gan H, Cao D, Kypridis A, Smyth FE, Brechbiel MW, Burgess AW, Old LJ, Scott AM (2005) Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR. Br J Cancer 92:1069–1077
Erbitux (cetuximab) [package insert] (2013) Indianapolis, IN: Eli Lilly
Tan AR, Moore DF, Hidalgo M, Doroshow JH, Poplin EA, Goodin S, Mauro D, Rubin EH (2006) Pharmacokinetics of cetuximab after administration of escalating single dosing and weekly fixed dosing in patients with solid tumors. Clin Cancer Res 12:6517–6522
Baselga J, Pfister D, Cooper MR, Cohen R, Burtness B, Bos M, D’Andrea G, Seidman A, Norton L, Gunnett K, Falcey J, Anderson V, Waksal H, Mendelsohn J (2000) Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J Clin Oncol 18:904–914
Rowinsky EK, Schwartz GH, Gollob JA, Thompson JA, Vogelzang NJ, Figlin R, Bukowski R, Haas N, Lockbaum P, Li YP, Arends R, Foon KA, Schwab G, Dutcher J (2004) Safety, pharmacokinetics, and activity of ABX-EGF, a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol 22:3003–3015
Hatanpaa KJ, Burma S, Zhao D, Habib AA (2010) Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 12:675–684
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN, Iafrate AJ (2011) Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20:810–817
Yasuda H, Kobayashi S, Costa DB (2012) EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 13:e23–e31
Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, Huberman MS, Cohen DW, Nakayama S, Ishioka K, Yamaguchi N, Hanna M, Oxnard GR, Lathan CS, Moran T, Sequist LV, Chaft JE, Riely GJ, Arcila ME, Soo RA, Meyerson M, Eck MJ, Kobayashi SS, Costa DB (2013) Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 5:216ra177
Men HT, Gou HF, Qiu M, He JP, Cheng K, Chen Y, Ge J, Liu JY (2014) A case of penile squamous cell carcinoma treated with a combination of antiepidermal growth factor receptor antibody and chemotherapy. Anticancer Drugs 25:123–125
Brown A, Ma Y, Danenberg K, Schuckman AK, Pinski JK, Pagliaro LC, Quinn DI, Dorff TB (2014) Epidermal growth factor receptor-targeted therapy in squamous cell carcinoma of the penis: a report of 3 cases. Urology 83:159–165
Chaux A, Munari E, Katz B, Sharma R, Lecksell K, Cubilla AL, Burnett AL, Netto GJ (2013) The epidermal growth factor receptor is frequently overexpressed in penile squamous cell carcinomas: a tissue microarray and digital image analysis study of 112 cases. Hum Pathol 44:2690–2695
Carthon BC, Ng CS, Pettaway CA, Pagliaro LC (2014) Epidermal growth factor receptor-targeted therapy in locally advanced or metastatic squamous cell carcinoma of the penis. BJU Int 113:871–877
Gou HF, Li X, Qiu M, Cheng K, Li LH, Dong H, Chen Y, Tang Y, Gao F, Zhao F, Men HT, Ge J, Su JM, Xu F, Bi F, Gao JJ, Liu JY (2013) Epidermal growth factor receptor (EGFR)-RAS signaling pathway in penile squamous cell carcinoma. PLoS One 8:e62175
Acknowledgments
Medical writing support was provided by Jacqueline Nielsen, an AbbVie Inc. employee.
Funding
This study was funded by AbbVie Inc.
Author contributions
All authors participated in the collection and analysis of data, manuscript writing or editing, and provided final approval of the manuscript.
Sponsor disclosures
The design, study conduct, analysis, and financial support of the clinical trial were provided by AbbVie Inc. AbbVie Inc. participated in the interpretation of data, review, and approval of this manuscript.
Compliance With Ethical Standards
ᅟ
Conflict of interest
J M Cleary, D A Reardon, N Azad, L Gandhi, G I Shapiro, and Jorge Chaves declare that they have no conflicts of interest. M Pedersen, P Ansell, W Ames, H Xiong, W Munasinghe, M Dudley, E Reilly, K Holen, and R Humerickhouse are employees of AbbVie Inc. and may own AbbVie Inc. stocks or options.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cleary, J.M., Reardon, D.A., Azad, N. et al. A phase 1 study of ABT-806 in subjects with advanced solid tumors. Invest New Drugs 33, 671–678 (2015). https://doi.org/10.1007/s10637-015-0234-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0234-6