
Summary
This study was designed to evaluate the efficacy, safety profile, pharmacokinetics, pharmacodynamics and quality of life of pegylated recombinant human arginase 1 (Peg-rhAgr1) in patients with advanced hepatocellular carcinoma (HCC). Patients were given weekly doses of Peg-rhAgr1 (1600 U/kg). Tumour response was assessed every 8 weeks using RECIST 1.1 and modified RECIST criteria. A total of 20 patients were recruited, of whom 15 were deemed evaluable for treatment efficacy. Eighteen patients (90 %) were hepatitis B carriers. Median age was 61.5 (range 30–75). Overall disease control rate was 13 %, with 2 of the 15 patients achieving stable disease for >8 weeks. The median progression-free survival (PFS) was 1.7 (95 % CI: 1.67–1.73) months, with median overall survival (OS) of all 20 enrolled patients being 5.2 (95 % CI: 3.3–12.0) months. PFS was significantly prolonged in patients with adequate arginine depletion (ADD) >2 months versus those who had ≤2 months of ADD (6.4 versus 1.7 months; p = 0.01). The majority of adverse events (AEs) were grade 1/2 non-hematological toxicities. Transient liver dysfunctions (25 %) were the most commonly reported serious AEs and likely due to disease progression. Pharmacokinetic and pharmacodynamic data showed that Peg-rhAgr1 induced rapid and sustained arginine depletion. The overall quality of life of the enrolled patients was well preserved. Peg-rhAgr1 is well tolerated with a good toxicity profile in patients with advanced HCC. A weekly dose of 1600 U/kg is sufficient to induce ADD. Significantly longer PFS times were recorded for patients who had ADD for >2 months.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The incidence of hepatocellular carcinoma (HCC) is rising worldwide, with over 500,000 new cases reported annually, the large majority of them being in Asia (notably Southern China) owing to the high prevalence of hepatitis B carriers [1]. Surgical resection and liver transplantation have been the mainstays of attempted curative treatment. However, <20 % of patients are suitable for surgery as most present with advanced disease and poor liver function [2].
HCC cells are generally auxotrophic for arginine since, unlike normal somatic cells, they cannot recycle arginine from citrulline, making them totally reliant on exogenous arginine for maintenance and growth. Thus, arginine deprivation therapy can potentially be beneficial in the treatment of HCC. In fact, enzymic removal of arginine, whether with pegylated recombinant human arginase 1 (Peg-rhAgr1) or arginine deiminase (ADI), is now a new platform of anti-cancer treatment in a number of human malignancies [3, 4], especially HCC [5–8]. Preclinical data shows that Peg-rhArg1 may synergise with systemic HCC chemotherapy, such as oxaliplatin, supporting its use in combinatorial therapy in the clinical setting [9].
Our phase 1 safety and dose-finding study of Peg-rhArg1 in advanced HCC clearly showed that arginine depletion in humans could be achieved safely, and with preliminary indication of clinical efficacy in those patients who had achieved adequate arginine depletion (ADD) [8], defined as the circulatory arginine level < 8 μM as long as the drug was administered. This gave an ADD dose of Peg-rhArg1 as 1600 U/kg per week as a basis for further clinical trials. The aim of this study was to evaluate the potential efficacy of Peg-rhArg1 and define its safety profile, pharmacokinetics/ pharmacodynamics and quality of life at 1600 U/kg in patients with advanced hepatocellular carcinoma.
Materials and methods
This was a prospective, single-center, open-label, single-arm phase II study of Peg-rhArg1 in subjects with advanced HCC. The protocol was approved by the local ethics committee and written consents were obtained from the patients before enrollment. It was registered in ClinicalTrials.gov (NCT01092091).
Patient eligibility
Advanced or metastatic HCC patients unsuitable for surgery or other loco-regional therapies were enrolled. HCC was diagnosed either by cyto-histological confirmation or by non-invasive criteria set by the European Association for the Study of Liver Disease: cirrhotic patients with either two coincident imaging techniques demonstrating focal lesion > 2 cm with arterial hypervascularization or one imaging technique showing similar arterial hypervascularization associated with an alpha fetal protein (AFP) level > 400 ng/ml [10]. Fine needle cytology and/or biopsy were used for histological confirmation when necessary. Staging was by both American Joint Committee on Cancer (AJCC) and Barcelona Clinical Liver Cancer (BCLC) staging systems. Other eligibility criteria included adult patients aged 18–75 years; patients with Child-Pugh class A or B cirrhosis; Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1; a life expectancy of ≥ 12 weeks with adequate organ function; complete blood picture (absolute neutrophil count ≥ 1.0 × 109/L, platelet count ≥ 100 × 109/L or INR ≤ 2.0) and biochemistry (total bilirubin ≤ 40 μmol/L, serum albumin ≥ 30 g/L, serum alanine transaminase ≤ 5 × upper limit of normal). The disease also had to be measurable with at least one lesion, which was >1 cm in one dimension either on computed tomography (CT) or magnetic resonance imaging (MRI) scans. Major exclusion criteria included advancing liver failure; significant cardiac or pulmonary disease defined by New York Heart Association Class III or IV, VEF < 50 % by echo or MUGA, or a history of myocardial infarction within the past 6 months, significant unstable arrhythmia or evidence of ischemia on electrocardiogram; significant active infection including HIV requiring oral or parenteral anti-infective therapies; prior treatment with arginine depleting agent.
Treatment design and dose modifications
The recruited patients received weekly intravenous doses of 1600 U/kg Peg-rhAgr1 alone. There was no dose adjustment. Toxicities were graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) v3.0 scale [11].
Disease evaluation
A full history and complete physical examination, including performance status, was carried out every 2 weeks during the initial clinic visits. Laboratory tests—blood counts, biochemistry and alpha fetal protein level - were also checked during every visit. Disease was assessed by CT scan every 8 weeks until it documented tumor progression. Magnetic resonance imaging and positron emission tomography with 11C-acetate as radiotracer were included in case of uncertainty of response. Response was classified according to both RECIST 1.1 and modified RECIST criteria [12]. After disease progression had been documented, patient survival was recorded every 2 months until death. Response in this study was classified as complete response (CR), partial response (PR), stable disease (SD) or progressive disease (PD). Patients who had achieved CR, PR or SD were defined as achieving disease control.
Efficacy and endpoints
The primary endpoint was disease control rate (DCR). The secondary endpoints were safety, progression free survival (PFS), overall survival (OS), pharmacokinetic and pharmacodynamic data, and quality of life (QOL) of the enrolled patients.
Pharmacokinetics (PK) and pharmacodynamics (PD) measurements
Plasma arginase and corresponding arginine levels were measured. Blood samples were drawn at baseline, 2 and 4 h after single dosing to establish maximum plasma concentration (Cmax) and initial clearance. Additional single time-point samples were drawn on days 2 and 4 to establish the terminal T1/2 and duration of arginine depletion after the first dose. Pre-dose samples and 24 h post-dose samples were obtained from weeks 2 and 3, i.e. on days 8, 9, 15 and 16; and pre-dose samples from week 4 onwards until the last day of treatment were also obtained when the patient was off-study when possible.
PK parameters were calculated based on a non-compartmental model approach which included: Cmax, time to maximum observed concentration (Tmax), minimum observed concentration (Cmin), and area under the curve (AUC∞). Arginase levels were measured using an ELISA Kit (ExCell Biology, China) per standard protocol. Arginine levels were measured using a high-speed amino acid analyser [13]. The ADD dose was defined as the amount of Peg-rhAgr1 that depletes arginine level to < 8 μM throughout the treatment period.
Safety monitoring
Safety analysis was based on an intention-to-treat (ITT) population. All patients who had been enrolled in the trial and received at least one dose of medication were included in the ITT population.
Safety assessments involved monitoring and recording all adverse events (AEs) and serious adverse events (SAEs) throughout the trial period. Apart from monitoring of vital signs, urinalysis, hematology and blood chemistry were followed. Patients who had received at least one dose of treatment were considered in the safety assessment. AEs and SAEs were classified according to NCI-CTCAE v3.0 [11].
Quality of life assessment
Health-related quality of life (HRQOL) was assessed in participating patients using the European Organization for Research and Treatment of Cancer quality of life questionnaire (EORTC QLQ C-30) version 1 and QLQ-HCC18. The HRQOL questionnaires were given by the research staff and filled in by the individual patients, and every effort was made to do this prior to consultations with the clinicians. Baseline HRQOL assessment was made before enrollment and at 4-weekly intervals thereafter until the end of treatment.
Statistical analysis
Statistical analyses were performed on an ITT basis using SAS® Software version 9.2 (SAS Institute, Cary, NC, USA). Survival analysis was computed by the Kaplan-Meier method. PFS was defined as the time from treatment to radiological progression or death due to any causes, or date of last contact if a patient that did not have disease progression died at the time of analysis. OS was defined as the time from treatment to the date of death due to any causes or last follow-up date.
Results
Demographics
Between March 2010 and February 2012, 20 subjects were enrolled. The median age was 61.5 (range: 30–75) years. All subjects were of Chinese descent and only one being female (Table 1). Eighteen (90 %) patients were hepatitis B virus (HBV) carriers. Anti-HBV medications such as entecavir (35 %) and lamivudine (15 %) were common concurrent medications at baseline. The majority of the enrolled patients had Child-Pugh A cirrhosis (90 %) and were in advanced disease stage at baseline. The study population was heavily pretreated. Fifteen patients had at least two rounds of trans-arterial chemoembolization, 2 of whom also had systemic biologics everolimus and sorafenib.
Efficacy
Fifteen patients completed more than 8 weeks of study and were evaluable for treatment efficacy and PFS. Five patients terminated the study prior to efficacy assessment due to the following reasons: 3 patients died of rapid disease progression, 1 patient stopped the trial medication due to pneumonia, and 1 patient terminated because of other unrelated medical conditions.
Among the evaluable subjects, the overall DCR was 13 % with two patients having stable disease after 8 weeks of treatment according to both RECIST 1.1 and modified-RECIST. No complete response or partial response was reported. Regarding AFP response, for those evaluable patients with baseline AFP level > 20 ng/ml (n = 9), 3 of them had a 4–10 % reduction in their log AFP levels during the trial. The median duration of drug exposure was 8 weeks (range: 1–32 weeks).
Survival analyses
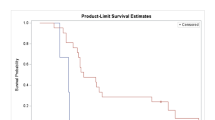
Among the ITT patients, the median PFS was 1.7 months (95 % confidence interval [CI]: 1.67–1.80; Figure 1). The median OS for the ITT population and evaluable patients was 5.2 (95 % CI: 3.3–12.0) and 6.4 months (95 % CI: 4.7–12.0; Figure 2), respectively. At the time of analysis, 5 patients were still alive and 15 patients had died. The majority of the deaths were attributed to progression of underlying HCC, except one patient who died of pneumonia.

Progression-free survival of evaluable patients

Overall Survival of all Enrolled Patients
Regarding the relationship of survival with ADD, there was a statistically significant difference of PFS in patients with ADD for > 2 months compared with that in patients with ADD duration ≤ 2 months (6.4 versus 1.7 months; p = 0.01). A difference in OS was found in the group of ITT patient having ADD duration < 1 month (median: 4.7 months) compared to those of 1–2 month duration (median: 5.2 months) and > 2 months (median: 7.4 months), although none reached significance with the small numbers involved (p = 0.72).
Adverse events and safety analyses
All enrolled patients were evaluated for AEs, summarized in Table 2 regardless of cause. Most of them were mild, grade 1 or 2 in severity, and not related to the trial medication.
A total of 17 SAEs were reported in the ITT population with liver dysfunction being the most common (3 patients; 15 %). Abdominal distention (10 %), abdominal pain (10 %), and tumor pain (10 %) were also common SAEs. None of these SAEs was directly linked to the enzyme treatment.
PK and PD analyses
Table 3 summarizes the PK parameters for 18 of 20 ITT subjects. Our pegylated drug had a relatively long half-life (mean: 105 h; range: 43 to 117 h). The inter-subject variability of clearance and volume of distribution were low with means of 0.16 U*ml/h*kg*μg (cv, 33 %) and 24.7 U*ml/h*kg*μg (cv, 25 %), respectively. There was little accumulative effect of Peg-rhArg1 for the first 3 weekly combination doses; the mean AUC 24 h post doses were kept between 1126 and 1373 h*μg/ml.
The optimal dose of Peg-rhArg1, i.e. 1600 U/kg per week, was able to reduce the plasma arginine level to below quantification limit (5 μM) effectively. The time to reach effective ADD was 2 h post dose for all patients, by which time Peg-rhArg1 had reached a plateau. There was also a long ADD duration (median 1 month; STD 49 days) effected by Peg-rhArg1. Median arginine levels remained below quantification limit after 8 weeks of Peg-rhArg1 treatment.
Quality of life analyses
The mean score of EORTC QLQ C-30 physical functioning scale decreased from 82.7 to 70.7 during the study period. In particular, the mean scores of role functioning, emotional functioning, cognitive functioning, social functioning, and financial difficulties had small, but non-significant, drops from screening to week 8. On the other hand, pain, insomnia, constipation, and loss of appetite increased slightly after 8 weeks of treatment. Fatigue, nausea and vomiting were relatively worse and their mean scores increased from 26.7 to 36.3 and 5.8 to 14.4, respectively. Of the total 15 scales, a significant difference (p = 0.02) between screening and week 8 assessments was only shown in the physical functioning scale. No significant changes from screening to week 8 were detected in the other C30 scales.
Regarding EORTC QLQ-HCC18, all of the mean scores increased between screening and week 8, except nutrition. Nevertheless, no significant difference was detected after 8 weeks treatment.
Discussion
As in our previous published phase I study by Yau et al. [8], we have demonstrated here that 1600 U/kg (2.7 mg/kg) of pegylated recombinant human arginase (Peg-rhArg1) per week is a suitable dose to give adequate depletion dose (ADD), i.e. < 8 μM. Although Peg-rhAgr1 exhibits some inter-subject pharmacodynamics variability, it is not only effective in depleting plasma arginine for a prolonged period (in one patient for as long as 32 weeks), but is safe, with very low toxicity and good quality of life assessments. In an average 60 kg subject, this 2.7 mg/kg of Peg-rhArg1 translates into 162 mg protein challenge per week, which is not high. Our current PK/PD data therefore support the weekly dosing schedule of Peg-rhAgr1 as safe and effective in inducing ADD for future clinical studies.
Of the 17 SAEs reported, none was hematological. The grade 3 and 4 toxicities, such as tumor pain, abdominal pain and distension and deranged liver function, were undoubtedly tumor related, due to disease progression. The QOL measurements with EORTC QLQC-30 between screening and week 8 assessments showed significant difference in physical functional scale only, with preservation of other C30 scales. Regarding EORTC QLQ-HCC18 analysis, all the mean scores had increased, except nutrition. Both physical functional and nutritional scores are going to be affected in patients with terminal cancer, and it would be difficult to attribute any of them to the study drug. It is also reasonable to conclude that these studies confirmed that QOL was well preserved during treatment.
Our analysis showed associations between ADD and disease response parameters; ADD duration correlated with OS: ADD < 1 month OS = 4.7 months; ADD 1–2 months OS = 5.2; ADD > 2 months OS = 7.4 months, which, although indicative of a positive trend, was not statistically significant. Of the patients with ADD > 2 months, a significantly longer PFS was achieved compared with those who had ADD < 2 months (median, 6.4 versus 1.7 months; p ≤ 0.05). The results show that the longer ADD duration can be achieved, the longer the OS and PFS; ADD should therefore be included as one of the response evaluation criteria in future clinical studies.
Our OS of 6.4 month in 15 evaluable cases (5.2 months in all 20 patients) is encouraging, considering untreated HCC would have a survival expectancy of <2–3 months [14]. This was achieved despite 75 % of the patients having been heavily pretreated with TACEs and chemotherapy. In the Sorafenib Asia-Pacific Study [15], in which majority (>70 %) of the subjects were HBV+ (our present report 90 %), patients in the treatment arm only had a meager OS of 6.5 months vs. 4.2 in the placebo. This OS duration similar to our own suggests Peg-rhArg1 may have survival advantage over sorafenib, considering all the patients in Sorafenib Asian Pacific Study had no prior treatment and were all Child-Pugh A with better treatment baseline. Kelley et al. analysed the 178 patients with HCV+ HCC in the Sorafenib registration study (SHARP) and found that the OS duration was in fact 14 months vs. 7.9 in the placebo group (16), which contrasts with the short OS of 6.5 months in HBV+ HCC treated with Sorafenib in the Asian Pacific Study (15). Therefore, there is still an unmet medical need for HBV-induced HCC in the present era of targeted therapy, which could be addressed by novel agents such as Peg-rhArg1, given its proven mild toxicity profile and potential synergy with chemotherapy such as oxaliplatin and 5 FU as reported in our preclinical models (9). This, we hope to demonstrate in our ongoing expanded phase II trial in which Peg-rhArgl is combined with oxaliplatin and capecitabine in patients with mainly HBV-induced HCC.
In the present study, no demonstrable anti-tumor response was shown—only two of the 15 evaluable cases had stable disease (SD) as assessed with RECIST criteria. It is also too early to draw any meaningful conclusion from this result because of the low numbers and heavy pre-treatment that the patients had; however, the findings seem consistent with the low response rates with any other target therapies used singly in the treatment of HCC, which includes the current standard, sorafenib. As single agent, sorafenib (SHARP Study) [16] has scant anti-tumor activity; it was given regulatory approval entirely on the basis of its OS and time to radiological progression (TTP) advantages (OS 10.7 months vs. 7.9 months, p < 0.01; TTP 5.5 months vs. 2.8 months, p < 0.001) over the placebo control. It should be stressed that this was only achieved at the expense of moderate to severe systemic toxicities, including grade 3 and 4 hand-foot skin reactions, diarrhea, hypertension and rash, none of which - by virtue of different drug mechanism - were not seen in Peg-rhArg1.
The favorable toxicity profile and tolerance to our drug should allow incorporation of other anti-HCC modalities, such as chemotherapy or target therapies, including tyrosine kinase inhibitors [15, 17], since this is decidedly the route towards a better clinical outcome for the HCC patient in particular. This is particularly pertinent in the treatment of advanced cases, since most patients have ongoing chronic hepatitis that may flare up at any time during treatment, liver cirrhosis, and compromised liver function, leading to intolerance to any form of treatment. China’s Food & Drug Administration (CFDA) recently announced that oxaliplatin-based chemotherapy (FOLFOX4) is one to be of the accepted treatments for HCC in an oriental setting (EACH Study) [18]. This has rekindled interest in the possible role of chemotherapy in the treatment of advanced HCC, hitherto thought to be highly resistant to systemic chemotherapy, although oncologists have reported low response rates in HCC treated with several chemotherapeutic agents, including platinoids and anti-metabolites (5-fluorouracil) [19]. In one single-armed phase II study, Cisplatin, Interferon, Adriamycin and 5-fluorouracil (PIAF) yielded partial response in 13/50 (26 %) patients [20]. The EACH Study was a randomized phase III study comparing FOLFOX4 with control Doxorubincin in a 1:1 fashion; this revealed tolerability and efficacy of FOLFOX4 in HCC compared with the control, with significant advantages in OS (6.47 versus 4.9 months, p = 0.0425), PFS (2.97 versus 1.8 months, p = 0.0003), DCR (53.26 % vs. 32.62, p = 0.0001) and RR (8.7 % vs. 2.7 %, p = 0.014). In preclinical models, Peg-rhArg1 synergizes with both oxaliplatin, hydroxyurea and 5FU in vitro, and has at least an additive effect in vivo in HCC xenografts [9, 21]. On the basis of the CFDA pronouncement and our preclinical experience, there is every good reason to test the efficacy of Peg-rhArg1 in combination with oxaliplatin and capecitabine (Xelox), as is currently ongoing in our expanded phase II study.
The randomized phase II pegylated deiminase (Peg-ADI) study in Asian [7] was carried to test the efficacies of two different dose levels of Peg-ADI. The median OS in subjects who had prolonged arginine depletion for >4 weeks was longer, though not statistically significant because of the wide variation in response, than those who had arginine depletion for <4 weeks (mean 10 versus 5.8 months; p = 0.251). The failure to achieve arginine depletion in 25 out of 61 patients in their study was due to antibody formation, probably related to PEG dissociation exposing the xenobiotic enzyme. This is not the case with Peg-rhArg1 and antibodies to Peg-rhArg1 have not been detected in the present study.
There are definite biomarkers in HCC that can be predictive and/or prognostic with Peg-rhArg1, i.e. the urea cycle enzymes argininosuccinate synthetase (ASS – coupled with argininosuccinate lyase ASL) and ornithine transcarbamoylase (OTC) [22–24, 5]. Absence or deficiency of these enzymes, whether at the transcription or translational level, prevents the synthesis (recycling) of arginine from their two precursors citrulline and ornithine, respectively. Selection of cases that could be more sensitive to ADD on the basis of ASS/ASL negativity would greatly increase the chances of Peg-rhArg1 treatment being beneficial, as a retrospective analysis of these biomarkers had indicated in the ADI Asian study [7]. In our current phase II studies in Hong Kong, we have mandated the incorporation of these biomarkers ab initio to test their correlation with response and prognosis to arginine depletion with Peg-rhArg1.
Arginine depletion, although currently not in the mainstream treatment in human malignancies, has emerged as a novel anti-cancer treatment. It is based on sound scientific rationale that has emerged from a large volume of published preclinical data [25–30]. Currently, there are two arginine depleting agents in clinical phase development worldwide, viz. Peg-ADI and Peg-rhArg1. From published clinical studies [7, 8], it appears that single agent activity, whether Peg-ADI or Peg-rhArg1, could, at most, induce disease stability or marginal OS advantage. However, with combination therapy, Peg-rhArg1 could potentially augment or synergize systemic chemotherapy or target therapies, and prove to be efficacious in a number of arginine-auxtrophic cancers, including certain leukemias, as noted in a recent MD Anderson study [31]. The authors of this report claimed that the combination arginase/cytarabine was more beneficial than single agent arginase in T-cell leukemia. Another possible advantage of arginine depletion with Peg-rhArg1 rather than peg-ADI is the problem of ammonia release with the latter. In patients with compromised liver function, the additional ammonia load can lead to hepatic encephalopathy.
The present study helps to dispel the impression that arginase is less favorable of the two enzymes, not warranting its place in clinical use by virtue of a high Km in the range of 2–5 mM and its apparent “suboptimal” enzymic activity at physiological pH [32]. While its specific activity might be lower, its high Kcat (426 sec-1) indicates that its rate of catabolism of arginine is fast, thereby compensating for its higher Km. This is evident from the rapid action on plasma arginine well within 2 h period of treatment. That the enzyme has higher activity at pH 9.1 than at pH 7.0–7.4 is a purely chemical observation and has little relevance, since arginase is a natural liver enzyme that works efficiently at physiological pH. This favorable PK/PD profile with our study drug may well be due to the small changes in the biochemical properties of the enzyme following pegylation, since the Km is virtually unchanged and the Kcat only compromised by about 15–20 %. The extent to which pegylation alters not only the half-life, the specific activity and immunogenicity of the two enzymes may not be that different, but these parameters have yet to undergo strict comparison. However, the xenobiotic nature of ADI, coupled with the toxic product of its catabolism of arginine (NH3), remain two of the strongest reasons for considering the advantages of the natural enzyme (arginase I) in cancer therapy.
References
Parkin DM, Bray F, Ferlay J, Pisani P (2001) Estimating the world cancer burden: Globocan 2000. Int J Cancer 94(2):153–156. doi:10.1002/ijc.1440
Poon RT, Fan ST, Tsang FH, Wong J (2002) Locoregional therapies for hepatocellular carcinoma: a critical review from the surgeon’s perspective. Ann Surg 235(4):466–486
Wheatley DN (2004) Controlling cancer by restricting arginine availability–arginine-catabolizing enzymes as anticancer agents. Anti-Cancer Drugs 15(9):825–833
Wheatley DN, Campbell E, Lai PB, Cheng PN (2005) A rational approach to the systemic treatment of cancer involving medium-term depletion of arginine. Gene Ther Mol Biol 9:33–40
Cheng PN, Lam TL, Lam WM, Tsui SM, Cheng AW, Lo WH, Leung YC (2007) Pegylated recombinant human arginase (rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res 67(1):309–317. doi:10.1158/0008-5472.CAN-06-1945
Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, Cremona F, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA, Ng C, Curley SA (2004) Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol 22(10):1815–1822. doi:10.1200/JCO.2004.11.120
Yang TS, Lu SN, Chao Y, Sheen IS, Lin CC, Wang TE, Chen SC, Wang JH, Liao LY, Thomson JA, Wang-Peng J, Chen PJ, Chen LT (2010) A randomised phase II study of pegylated arginine deiminase (ADI-PEG 20) in Asian advanced hepatocellular carcinoma patients. Br J Cancer 103(7):954–960. doi:10.1038/sj.bjc.6605856
Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, Pang R, Fan ST, Poon RT (2013) A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs 31(1):99–107. doi:10.1007/s10637-012-9807-9
Chow AK, Ng L, Sing Li H, Cheng CW, Lam CS, Yau TC, Cheng PN, Fan ST, Poon RT, Pang RW (2012) Anti-tumor efficacy of a recombinant human arginase in human hepatocellular carcinoma. Curr Cancer Drug Targets 12(9):1233–1243
Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, Christensen E, Pagliaro L, Colombo M, Rodes J (2001) Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol 35(3):421–430
Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN, Rubin P (2003) CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol 13(3):176–181. doi:10.1016/S1053-4296(03)00031-6
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Cheng PN, Leung YC, Lo WH, Tsui SM, Lam KC (2005) Remission of hepatocellular carcinoma with arginine depletion induced by systemic release of endogenous hepatic arginase due to transhepatic arterial embolisation, augmented by high-dose insulin: arginase as a potential drug candidate for hepatocellular carcinoma. Cancer Lett 224(1):67–80. doi:10.1016/j.canlet.2004.10.050
Yau T, Yao TJ, Chan P, Ng K, Fan ST, Poon RT (2008) A new prognostic score system in patients with advanced hepatocellular carcinoma not amendable to locoregional therapy: implication for patient selection in systemic therapy trials. Cancer 113(10):2742–2751. doi:10.1002/cncr.23878
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z (2009) Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10(1):25–34. doi:10.1016/S1470-2045(08)70285-7
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359(4):378–390
Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, Chung HC, Song X, Xu J, Poggi G, Omata M, Pitman Lowenthal S, Lanzalone S, Yang L, Lechuga MJ, Raymond E (2013) Sunitinib versus Sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol 31(32):4067–4075. doi:10.1200/JCO.2012.45.8372
Qin S, Bai Y, Lim HY, Thongprasert S, Chao Y, Fan J, Yang TS, Bhudhisawasdi V, Kang WK, Zhou Y, Lee JH, Sun Y (2013) Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol 31(28):3501–3508. doi:10.1200/JCO.2012.44.5643
Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, Hui P, Ma B, Lam KC, Ho WM, Wong HT, Tang A, Johnson PJ (2005) A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst 97(20):1532–1538. doi:10.1093/jnci/dji315
Leung TW, Patt YZ, Lau WY, Ho SK, Yu SC, Chan AT, Mok TS, Yeo W, Liew CT, Leung NW, Tang AM, Johnson PJ (1999) Complete pathological remission is possible with systemic combination chemotherapy for inoperable hepatocellular carcinoma. Clin Cancer Res: Off J Am Assoc Cancer Res 5(7):1676–1681
Wheatley DN (2012) Amino acid deprivation as the primary intervention in cancer therapy: cellular basis of effective combinatorial therapy with arginase as the platform. Biomed Res 23:149–154
Bowles TL, Kim R, Galante J, Parsons CM, Virudachalam S, Kung HJ, Bold RJ (2008) Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int J Cancer 123(8):1950–1955. doi:10.1002/ijc.23723
Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA (2004) Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer 100(4):826–833. doi:10.1002/cncr.20057
Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH, Kim JH, Park IS, Yoon DK, Min BH (2007) Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int J Cancer 120(4):897–905. doi:10.1002/ijc.22322
Hsueh EC, Knebel SM, Lo WH, Leung YC, Cheng PN, Hsueh CT (2012) Deprivation of arginine by recombinant human arginase in prostate cancer cells. J Hematol Oncol 5:17. doi:10.1186/1756-8722-5-17
Lam TL, Wong GK, Chong HC, Cheng PN, Choi SC, Chow TL, Kwok SY, Poon RT, Wheatley DN, Lo WH, Leung YC (2009) Recombinant human arginase inhibits proliferation of human hepatocellular carcinoma by inducing cell cycle arrest. Cancer Lett 277(1):91–100. doi:10.1016/j.canlet.2008.11.031
Lam TL, Wong GK, Chow HY, Chong HC, Chow TL, Kwok SY, Cheng PN, Wheatley DN, Lo WH, Leung YC (2011) Recombinant human arginase inhibits the in vitro and in vivo proliferation of human melanoma by inducing cell cycle arrest and apoptosis. Pigment Cell Melanoma Res 24(2):366–376. doi:10.1111/j.1755-148X.2010.00798.x
Sugimura K, Ohno T, Kusuyama T, Azuma I (1992) High sensitivity of human melanoma cell lines to the growth inhibitory activity of mycoplasmal arginine deiminase in vitro. Melanoma Res 2(3):191–196
Takaku H, Takase M, Abe S, Hayashi H, Miyazaki K (1992) In vivo anti-tumor activity of arginine deiminase purified from Mycoplasma arginini. Int J Cancer 51(2):244–249
Tsui SM, Lam WM, Lam TL, Chong HC, So PK, Kwok SY, Arnold S, Cheng PN, Wheatley DN, Lo WH, Leung YC (2009) Pegylated derivatives of recombinant human arginase (rhArg1) for sustained in vivo activity in cancer therapy: preparation, characterization and analysis of their pharmacodynamics in vivo and in vitro and action upon hepatocellular carcinoma cell (HCC). Cancer Cell Int 9:9. doi:10.1186/1475-2867-9-9
Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, Cole J, Rodriguez PC (2010) Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood 115(25):5214–5221. doi:10.1182/blood-2009-12-258822
Dillon BJ, Holtsberg FW, Ensor CM, Bomalaski JS, Clark MA (2002) Biochemical characterization of the arginine degrading enzymes arginase and arginine deiminase and their effect on nitric oxide production. Med Sci Monit: Int Med J Exp Clin Res 8(7):BR248–BR253
Financial Support
Bio-Cancer Treatment International Limited and the University of Hong Kong Hepatocellular Carcinoma Research Grant.
Potential conflicts of interest
Drs Thomas Yau, Pierre Chan, Jimmy Yuen, Roberta Pang, Professor Sheung Tat Fan and Professor Ronnie Poon report no potential conflict of interest.
Dr Paul Cheng is the chief executive officer of Bio-Cancer Treatment International Limited. Dr Li Chen is the chief technical officer of Bio-Cancer Treatment International Limited. Professor Denys Wheatley is the chief scientific officer of Bio-Cancer Treatment International Limited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yau, T., Cheng, P.N., Chan, P. et al. Preliminary efficacy, safety, pharmacokinetics, pharmacodynamics and quality of life study of pegylated recombinant human arginase 1 in patients with advanced hepatocellular carcinoma. Invest New Drugs 33, 496–504 (2015). https://doi.org/10.1007/s10637-014-0200-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0200-8