
Summary
Introduction Cabazitaxel is a second-generation taxane with in vivo activity against taxane-sensitive and -resistant tumor cell lines and tumor xenografts. Cabazitaxel/cisplatin have therapeutic synergism in tumor-bearing mice, providing a rationale for assessing this combination in patients with solid tumors. Methods The primary objectives of this study were to determine dose-limiting toxicities (DLTs) and the maximum tolerated dose (MTD) of a cabazitaxel/cisplatin combined regimen (Part 1) and to assess antitumor activity at the MTD (Part 2). Safety and pharmacokinetics (PK) were also examined. Results Twenty-five patients with advanced solid tumors were enrolled (10 in Part 1; 15 in Part 2). In Part 1, two dose levels were evaluated; the MTD for cabazitaxel/cisplatin (given Q3W) was 15/75 mg/m2. DLTs occurring during Cycle 1 at the maximum administered dose (20/75 mg/m2; acute renal failure and febrile neutropenia) and the MTD (febrile neutropenia and hypersensitivity despite pre-medication) were as expected for taxane/platinum combinations. For the 18 patients treated at the MTD, the most frequent possibly related non-hematologic treatment-emergent adverse events (Grade ≥3) were nausea (16.7 %), fatigue, acute renal failure and decreased appetite (each 11.1 %). Neutropenia was the most frequent treatment-emergent Grade ≥3 hematologic laboratory abnormality at the MTD (77.8 %). The best overall response at the MTD was stable disease, observed in 66.7 % of patients. PK results of the combination did not appear to differ from single-agent administration for each agent. Conclusion Combination treatment with cabazitaxel/cisplatin had a manageable safety profile; no PK interactions were evident. The recommended Phase II dose for this combination is cabazitaxel/cisplatin 15/75 mg/m2 administered every 3 weeks. Antitumor activity findings suggest that further evaluation of this combination in disease-specific trials is warranted.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cabazitaxel is a second-generation taxane that is active against a broad spectrum of docetaxel-sensitive and docetaxel-resistant tumor cell lines and tumor xenografts [1]. Compared with docetaxel, cabazitaxel exhibits greater activity and a broader cytotoxicity profile against murine and human tumor cell lines. Furthermore, cabazitaxel has antitumor effects in multiple tumor xenograft models with acquired or innate resistance to docetaxel [1, 2]. Unlike docetaxel, cabazitaxel crosses the blood–brain barrier in rodents (showing activity in intracranial human glioblastoma models), leading to brain:plasma and brain:blood exposure ratios up to 6.51 and 9.02, respectively, after administration of 30 mg/kg cabazitaxel [2].
Cisplatin is a platinum-containing chemotherapeutic that interacts with DNA, creating DNA adducts that activate signal transduction pathways, ultimately resulting in apoptosis [3]. Preclinical studies have shown therapeutic synergism between cabazitaxel and cisplatin in tumor-bearing mice [4], and clinical studies of taxanes in combination with cisplatin have reported promising results [5–8]. As such, a combined cabazitaxel/cisplatin regimen could provide increased clinical benefits to patients with various solid tumors. Here, we report the results of a Phase I study of cabazitaxel/cisplatin treatment, which was designed to evaluate dose-limiting toxicities (DLTs), determine the maximum tolerated dose (MTD), assess potential antitumor activity, and examine safety and pharmacokinetics (PK).
Materials and methods
Study design
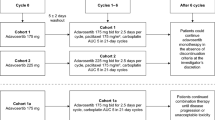
This was a Phase I, multicenter, open-label, single-arm, dose-escalation, two-part study, which included an expansion cohort to further evaluate the MTD. The primary objectives of Part 1 were to determine DLTs and the MTD of cabazitaxel administered in combination with cisplatin as a 1-h intravenous (IV) infusion every 3 weeks in patients with advanced solid tumors. Once the MTD had been established, the primary objective of Part 2 was to evaluate antitumor activity at the MTD. Secondary objectives included the examination of safety and PK (Parts 1 and 2), and to evaluate any drug–drug interaction between cabazitaxel and cisplatin (Part 1).
In Part 1, doses were escalated according to a standard 3 + 3 design (based on the safety evaluation [DLTs] during Cycle 1), with cohorts of 3–6 patients receiving study treatment on Day 1 every 3 weeks. The starting dose level (dose level 0) was cabazitaxel/cisplatin 20/75 mg/m2. Planned dose levels 1 and –1 for cabazitaxel/cisplatin were 25/75 mg/m2 and 15/75 mg/m2, respectively. The MTD was defined as the highest dose at which none of three or one of six patients evaluable for DLT experienced a DLT during the first treatment cycle. The maximum administered dose (MAD) was defined as the dose level at which at least two patients evaluable for DLT developed a DLT during the first treatment cycle.
In Part 2, additional patients were enrolled to investigate the safety, PK and antitumor activity of study treatment administered at the MTD (as determined in Part 1).
The study protocol and all amendments were approved by Institutional Review Boards and Independent Ethics Committees at participating institutions. All patients gave written informed consent. The study was conducted according to good clinical practice and the Declaration of Helsinki and its amendments. The trial was registered at www.ClinicalTrials.gov as NCT00925743.
Dose-limiting toxicities
A patient was evaluable for DLTs if they received any component of study treatment and had a DLT assessment during the first treatment cycle of Part 1. Patients who experienced a DLT could receive a lower dose of study treatment during later treatment cycles (after experiencing a DLT) at the investigator’s discretion.
DLTs were graded according to NCI-CTCAE v3.0 [9] and were defined as the following study treatment-related toxicities occurring during the first treatment cycle: non-hematologic toxicity of Grade ≥3 (except for Grade 3: fever without documented infection, inadequately treated nausea/vomiting/diarrhea/mucositis/stomatitis, fatigue, anorexia, elevated liver function tests that returned to baseline before the next treatment cycle, hypersensitivity reaction if the required pre-medication was not administered, or peripheral neuropathy that returned to Grade ≤1 before the next cycle); hematologic toxicity consisting of febrile neutropenia (fever ≥38.5 °C of unknown origin without clinically or microbiologically documented infection combined with Grade 3/4 neutropenia), Grade 4 neutropenia lasting >7 days or Grade 4 thrombocytopenia; or any other life-threatening toxicity.
Patient population
Adult patients with a histologically or cytologically confirmed metastatic or unresectable advanced solid malignancy, for which standard curative measures did not exist, but for which cisplatin-based therapy was considered appropriate, were eligible for this study. Prior taxane therapy was permitted in both Part 1 and Part 2. Exclusion criteria included: Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≥2; prior cabazitaxel treatment within 2 years or cisplatin treatment within 6 months; history of hypersensitivity to taxanes, cisplatin, polysorbate-80 or derivatives; history of significant hearing impairment; any clinically significant toxic effect (excluding alopecia) of prior therapy that had not resolved to Grade ≤1; prior chemotherapy, biologic therapy, targeted non-cytotoxic therapy, or radiotherapy within 3 weeks before registration; absolute neutrophil count <1,500/mm3, platelets <75,000/mm3, hemoglobin <9.0 g/dL or prothrombin time/international normalized ratio >1.5; estimated creatinine clearance <60 mL/min, or serum creatinine >1.0× upper limit of normal (ULN); total bilirubin > ULN, alkaline phosphatase >5.0× ULN, serum aspartate aminotransferase (AST) or serum alanine aminotransferase (ALT) >2.5× ULN if alkaline phosphatase is ≤2.5× ULN, or AST or ALT >1.5× ULN if alkaline phosphatase is >2.5× ULN and ≤5.0× ULN; or concurrent or planned treatment with a strong CYP3A4 inhibitor. For Part 2, patients with non-measurable disease were ineligible.
Study treatment
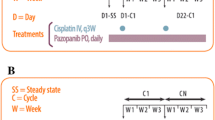
On Day 1 of each 3-week cycle, patients received a 1-h IV infusion of cisplatin followed by a 1-h IV infusion of cabazitaxel. At least 30 min before cabazitaxel was administered, patients received IV pre-medication with an antihistamine, a steroid and a H2 receptor antagonist. Antiemetic prophylaxis with aprepitant only was mandatory in Part 2, Cycle 2, while ondansetron, granisetron or dolasetron could be administered as needed in Part 1 (all cycles) and in cycles other than Cycle 2 in Part 2. For safety reasons, the dose of cabazitaxel was adjusted to a maximum body surface area (BSA) of 2.1 m2. Prophylactic use of granulocyte colony-stimulating factor (G-CSF) was allowed after Cycle 1, and therapeutic G-CSF was permitted during Cycle 1 following a hematologic DLT only. If G-CSF was administered outside of these parameters, the patient was not evaluable for DLTs.
New cycles of study treatment were not initiated until the absolute neutrophil count was ≥1,500/mm3, platelet count was ≥75,000/mm3, estimated creatinine clearance was ≥40 mL/min (in patients who had not received a prior cisplatin dose reduction for renal toxicity) or ≥50 mL/min (in patients who had received one cisplatin dose reduction for renal toxicity), liver function tests were within the range stipulated for study eligibility and non-hematologic toxicities (except alopecia, asthenia, local reactions, and other non-serious toxicities) had recovered to Grade ≤1 or baseline levels. Cycle length could be extended by a maximum of 2 weeks if additional time was required for resolution of study treatment-related toxicities or other treatment-emergent adverse events (TEAEs) regardless of causality.
In Part 1, dose reductions (except for those related to DLTs) were not permitted. In Part 2, up to two dose reductions were permitted for any patient with a significant toxicity. Dose re-escalations were not allowed.
Study treatment continued until disease progression, unacceptable toxicity, patient refusal of further treatment or investigator’s decision.
Safety assessments
Safety was assessed by medical history, concomitant medications, TEAEs, serious adverse events (SAEs), vital signs, physical examinations, ophthalmologic examinations, ECOG PS, electrocardiograms and laboratory safety tests (including complete blood count, coagulation, serum chemistry and urinalysis), both before study treatment and at designated intervals throughout the study. A TEAE was defined as any AE (regardless of relationship to study treatment) that developed during treatment or worsened in severity during treatment compared with baseline. A SAE was defined as any untoward medical occurrence at any dose that resulted in death, was life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability/incapacity, resulted in a congenital anomaly/birth defect or was a medically important event. All SAEs considered related to the study treatment were recorded throughout the study, regardless of when they occurred. Laboratory, vital signs and/or electrocardiogram abnormalities were recorded as TEAEs only if they were symptomatic, required corrective treatment, led to study treatment discontinuation and/or dose modification, and/or fulfilled a SAE criterion.
Pharmacokinetic assessments
To determine cabazitaxel plasma concentrations, blood samples were collected on lithium heparinate 5 min before the end of infusion, 5 and 15 min after the end of infusion, and 1.5, 2, 4, 6, 8, 9–11, 24 (1 day), 48 (2 days), 72 (3 days), 96 (4 days) – 120 (5 days) and 168 (7 days) hours after starting the infusion. Within 30 min of collection, samples were centrifuged at 2,000 g for 15 min at 4 °C to obtain the plasma fraction, which was stored at –20 °C until analysis. Cabazitaxel was extracted from plasma using an Oasis® HLB μElution plate. The quantitative determination was performed using a validated liquid chromatography–tandem mass spectrometry (LC/MS/MS) method [10], slightly modified using d6-cabazitaxel as an internal standard. The lower limit of quantification (LLOQ) was 1 ng/mL for a 100 μL sample size. Over the study period, the quality of the analytical method established by the coefficients of variation (CV) of the quality controls (for mean day-to-day variability of the estimated concentrations) showed a precision of ≤5.5 %. The assay accuracy, defined as the percentage difference between the nominal and the mean measured concentrations of quality controls, ranged from –7.67 to 2.75 %. For cisplatin, blood samples were collected on sodium heparinate immediately prior to infusion, 5 min before the end of infusion and at 1.5, 2, 3, 4, 6, 8, 11 and 24 h after starting the infusion. Within 20 min of collection, plasma was separated by centrifugation at 1,000 g for 10 min at 4 °C. Ultrafiltrate was generated by plasma ultracentrifugation at 2,000 g for 30 min at 4 °C in a Centrifree® device with an Amicon YM-30 ultrafiltration disc (Merck Millipore, Billerica, MA, USA), within 1 h of blood collection and stored at –20 °C before analyses.
The quantitative determination was validated following FDA guidance, and conducted using an inductively coupled plasma MS method after alkaline digestion. Before analysis, 100 μL samples (plasma or plasma ultrafiltrate, respectively) were digested for 30 min with either 9.9 mL internal standard (iridium) in 1 % TMAH/1 % EDTA for total platinum assay or with 0.9 mL internal standard in 1 % TMAH/1 % EDTA for ultrafiltrable platinum assay. The limit of quantitation was 100 ng/mL for total plasma platinum assay and 1 ng/mL for ultrafiltrable platinum assay. Over the study period, the analytical method showed a precision of ≤2.93 % (total platinum) and ≤6.09 % (ultrafiltrable platinum), and accuracy ranged from –2.10 to 3.2 % (total platinum) and from –0.30 to 1.20 % (ultrafiltrable platinum).
PK parameters were calculated from cabazitaxel plasma concentrations using non-compartmental analysis and were summarized by arithmetic mean, geometric mean, standard deviation, standard error of the mean, CV (%), minimum, median, maximum and number of observations with validated software (PKDMS version 2, running with WinNonlin Professional, version 5.2.1, Pharsight). PK parameters included: maximum plasma concentration observed (Cmax), first time to reach Cmax (Tmax), terminal half-life associated with the terminal slope (λz), area under the plasma concentration versus time curve calculated using the trapezoidal method from time 0 to the last measurable concentration (AUClast), area under the plasma concentration versus time curve extrapolated to infinity (AUC), total body clearance (CL) and volume of distribution at steady state (Vss). CL and Vss were also normalized using BSA. For cisplatin (total and ultrafiltrable platinum), Cmax, Tmax, and AUC0–24 were calculated using non-compartmental analysis using the same methodology as that for cabazitaxel.
Antitumor activity
Tumor response was evaluated using computed tomography or magnetic resonance imaging according to RECIST (version 1.1) [11]. Measurable target lesions, measurable non-target lesions and non-measurable non-target lesions were evaluated at screening, every two cycles (approximately every 6 weeks) thereafter, whenever disease progression was suspected, and at the end of study treatment or withdrawal, using the same method for each assessment. Bone scans were performed at baseline, whenever new or worsening bone symptoms occurred, and when tumor assessments were performed to confirm a response. Patients who discontinued study treatment before disease progression occurred continued to have tumor assessments every 6 weeks until disease progression or start of another anticancer therapy. The primary antitumor activity variable was the objective response rate (ORR), defined as the proportion of patients with confirmed complete response or partial response during the treatment period relative to the total number of patients in the analysis population. Time to progression and duration of response were also assessed.
Analysis populations
The all-treated (AT) population was defined as all enrolled patients who received at least one part of a dose of study treatment. The per-protocol (PP) population was defined as patients who received a minimum of two cycles of study treatment or withdrew early following progression/death due to progression within the same period, and who had a valid baseline tumor assessment, at least one post-baseline tumor assessment, and no major protocol deviation(s).
Safety was analyzed in the AT population. Antitumor activity (ORR) was analyzed in both the AT and PP populations. Patients who received the MTD in Part 1 were included in the antitumor activity evaluation in Part 2 of the study, provided they met the Part 2 eligibility criteria. The PK analysis was performed on patients in the AT population who had no major protocol deviation(s) and for whom any PK parameter was available.
Results
Patients
Twenty-five patients were enrolled (Table 1). The median age was 56 years and most patients had an ECOG PS of 1 (n = 15, 60 %). The most frequent primary tumor site was the lungs (n = 4, 16 %). At study entry, 8 % of patients had locally advanced disease and 92 % had metastatic disease. The most frequently involved organs at baseline were lymph nodes (80 %), lungs (56 %) and liver (40 %). At baseline, prior anticancer therapy, radiotherapy or surgery had been administered to 92 %, 60 % and 60 % of patients, respectively. Prior to this study patients had received a median of five different individual anticancer therapies (range: 2–9) and a median of 3.5 different anticancer regimens (including combinations; range: 1–8). A total of 14 patients (56 %) had received prior taxane treatment and six patients (24 %) had received prior cisplatin treatment.
Study treatment
During the dose-escalation phase (Part 1), 10 patients were treated at one of two dose levels: seven at dose level 0 (cabazitaxel/cisplatin doses of 20/75 mg/m2; MAD) and three at dose level –1 (15/75 mg/m2), which was found to be the MTD. During the cohort-expansion phase (Part 2), 15 patients were treated at the MTD. In total, 18 patients were treated at the MTD in the study. In Parts 1 and 2, 100 cycles of study treatment were administered: 40 at the 20/75 mg/m2 dose level and 60 at the 15/75 mg/m2 dose level. Three patients at 15 mg/m2 and two patients at 20 mg/m2 had their cabazitaxel doses capped in Cycle 1 and/or Cycle 2 due to BSA >2.1 m2. Median number of cycles of treatment administered per patient was three (range 1–8) with 15/75 mg/m2 and six (range 1–12) with 20/75 mg/m2. Median relative dose intensity for cabazitaxel at the 15/75 mg/m2 dose level was 0.954 (range 0.74–1.02), for cabazitaxel at the 20/75 mg/m2 dose level was 0.837 (range 0.57–0.96), for cisplatin at the 15/75 mg/m2 dose level was 0.988 (range 0.71–1.01) and for cisplatin at the 20/75 mg/m2 dose level was 0.856 (range 0.60–0.96). Three patients (14.3 %) received a reduced dose of cabazitaxel (one patient [6.7 %] who started at the 15/75 mg/m2 dose level and two patients [33.3 %] who started at the 20/75 mg/m2 dose level). Five patients (23.8 %) received a reduced dose of cisplatin (four patients [26.7 %] who started at the 15/75 mg/m2 dose level and one patient [16.7 %] who started at the 20/75 mg/m2 dose level) (Table 2). All patients discontinued study treatment. For patients who received the MTD, median duration of treatment was 10.1 weeks (range 3–26 weeks). Overall, disease progression was the most frequently reported reason for discontinuing study treatment (12 patients [48 %]) followed by TEAE occurrence (nine patients [36 %]) and withdrawal of consent or poor PS (two patients [8 %] each).
Dose escalations
Of nine patients evaluable for DLTs in Part 1, two experienced a DLT (Grade 3 acute renal failure and Grade 4 febrile neutropenia, both at the 20/75 mg/m2 dose level). Because no patient treated at 15/75 mg/m2 experienced a DLT in Part 1, this dose level was established as the MTD. Two of the 15 patients treated at the MTD in Part 2 experienced a DLT (Grade 3 febrile neutropenia and Grade 3 hypersensitivity despite required pre-medication). The 20/75 mg/m2 dose level was the MAD.
Safety
Safety data are summarized in Table 3. All patients experienced a TEAE; at the MTD, the most frequent all-grade non-hematologic TEAEs (regardless of relationship to study treatment) were nausea (77.8 %), vomiting (72.2 %), decreased appetite (66.7 %), fatigue (61.1 %) and diarrhea (44.4 %). All patients experienced a TEAE (all grades) considered possibly related to study treatment; at the MTD, the most frequent non-hematologic events were nausea (72.2 %), decreased appetite, vomiting (both 66.7 %) and diarrhea (44.4 %). At the MTD, nine patients (50.0 %) experienced a Grade ≥ 3 TEAE possibly related to study treatment; the most frequent non-hematologic events were nausea (16.7 %), decreased appetite, fatigue and acute renal failure (each 11.1 %). Sixteen (64.0 %) patients experienced a SAE (all grades and regardless of relationship to study treatment).
Patients experiencing hematologic and biochemical abnormalities (based on laboratory values) during study treatment are presented in Table 3. Neutropenia was the most frequently reported treatment-emergent Grade ≥ 3 hematologic abnormality, both overall (84.0 %) and at the MTD (77.8 %). Febrile neutropenia was observed in three patients (12.0 %) overall, including one patient at the MTD and two patients at 20/75 mg/m2.
Nine patients (36.0 %) experienced a TEAE (all grades) that led to study treatment discontinuation. At the MTD, four (22.2 %) patients developed a TEAE that led to study treatment discontinuation, including increased blood creatinine (11.1 %), increased blood urea and drug hypersensitivity (5.6 % each). Nine patients (36.0 %) had a study treatment delay due to a TEAE (all grades). At the MTD, five (27.8 %) patients had study treatment delayed due to TEAEs, including two (11.1 %) patients who developed anemia. Seven patients (28.0 %) had a dose reduction due to a TEAE (all grades). At the MTD, five (27.8 %) patients had a dose reduction due to TEAEs, including two (11.1 %) patients who developed increased blood creatinine.
Eleven patients (44 %) died during the study (eight who received the MTD and three who received the MAD). Causes of death were disease progression in nine patients, TEAE in one patient (septic shock after five treatment cycles at the MAD; death occurred 9 days after the last dose of study treatment) and unknown in one patient.
Pharmacokinetics
Plasma samples were collected from 20 patients, including four patients treated with 20/75 mg/m2 in Part 1 of the study and 16 patients treated with 15/75 mg/m2 (three patients in Part 1 and 13 patients in Part 2). Seven patients were excluded from the PK analyses; PK results for the 13 patients included in the statistical analysis are summarized in Tables 4 and 5. Overall, cabazitaxel CL normalized to BSA was 39.3 L/h/m2 (with moderate variability [CV 52 %]); CL was 39.7 L/h/m2 at 15/75 mg/m2 (n = 11) and 37.4 L/h/m2 at 20/75 mg/m2 (n = 2). Overall, cabazitaxel Vss normalized to BSA was 2,780 L/m2 with moderate variability (CV 35 %); Vss was 2,610 L/m2 at 15/75 mg/m2 and 3,730 L/m2 at 20/75 mg/m2. The PK of total and ultrafiltrable platinum was comparable across all dose levels. These results were comparable to prior PK studies of cabazitaxel.
Antitumor activity
Fifteen patients were evaluable for response, including three who received the MTD in Part 1. The best overall response was stable disease (10 patients, 66.7 %), which was observed in a range of tumor types: pancreas, lung (two patients each), prostate, pleura, endometrium, esophagus, ovary and ependyma (one patient each). Two patients had stable disease for six treatment cycles (the primary tumor sites were ovary and ependyma). Of the 15 patients evaluable for time to progression analysis, eight (53.3 %) progressed and seven (46.7 %) were censored; median progression-free survival was 2.7 months (95 % confidence interval 1.54–not determined) (Fig. 1).

Kaplan Meier analysis of progression-free survival in the per-protocol population (patients from Parts 1 and 2 treated at the MTD)
Discussion
Cabazitaxel and cisplatin are established cytotoxic agents with different mechanisms of action [3, 12]. Here, we aimed to establish the appropriate dosage and to assess the value of combination treatment of cabazitaxel plus cisplatin in patients with advanced solid tumors in terms of safety, PK and antitumor activity.
Based on available safety and dosing data from prior and ongoing clinical studies of cabazitaxel when the study was being designed, and the fact that cisplatin doses between 50 and 100 mg/m2 are feasible, a dosing schedule of cabazitaxel 20 mg/m2 and cisplatin 75 mg/m2 every 3 weeks was established as the starting dose for this study. Of the six evaluable patients treated at the MAD, two experienced a DLT (Grade 3 acute renal failure and Grade 4 febrile neutropenia). Therefore, the MTD and recommended Phase II dose of cabazitaxel administered in combination with 75 mg/m2 cisplatin on Day 1 of each 3-week cycle was found to be 15 mg/m2. These DLTs are consistent with those expected for a taxane/platinum agent combination regimen [13, 14]. The most frequently reported all-grade treatment-related TEAEs at the MTD were nausea, decreased appetite, vomiting and diarrhea; these AEs are also consistent with those of other taxane/platinum combinations [15, 16]. Of note, there was no suggestion that combining cabazitaxel with cisplatin resulted in increased peripheral neurotoxicity [17]. Neutropenia was the most frequently reported treatment-emergent Grade ≥3 hematologic abnormality (77.8 % of patients treated at the MTD). Because prophylactic use of hematopoietic growth factors was not permitted in the first treatment cycle, hematological tolerability may be improved in future studies and daily practice by using G-CSF.
The best tumor response was stable disease. In this study, patients were heavily pretreated with 92 % of patients having received prior chemotherapy and 56 % of patients having received prior taxane therapy; this may in part account for the limited antitumor activity that was observed. It is important to note that because no more than two evaluable patients had the same tumor type, it is not possible to draw any conclusions regarding antitumor activity in a given tumor type. Studies investigating taxane/cisplatin combinations have shown evidence of antitumor activity in both non-small cell lung cancer and nasopharyngeal cancer [15, 16], with ORRs of 47 % and 70.2 % reported, respectively. These studies provide evidence that taxane/platinum combinations are feasible and efficacious and as such, it may be prudent to conduct further studies in patients with specific cancers to determine the patient population in which the combination of cabazitaxel and cisplatin is most effective.
Cabazitaxel is extensively metabolized in the liver (>95 %), predominantly by the CYP3A isoenzyme (80–90 %) [18]. Cisplatin is not a known CYP substrate, inhibitor or inducer [19], therefore, no drug–drug interaction was expected. PK results observed in the small data set reported here are consistent with a lack of interaction between cabazitaxel and cisplatin at the doses investigated, supporting the potential feasibility of combining these drugs for the treatment of advanced solid tumors. Indeed, the CL of cabazitaxel administered with cisplatin 75 mg/m2 was within the range observed for cabazitaxel administered as monotherapy in patients with advanced solid tumor [10, 20, 21]. These results concur with a separate study that found that co-administration of cisplatin with a taxane did not alter the PK profile of the taxane [22]. In addition, the PK parameters of cisplatin (total and ultrafiltrable platinum) were comparable to those observed with single-agent administration [23].
In summary, the use of cabazitaxel in combination with cisplatin appears to be feasible, based on the small number of patients evaluated here. While these results would require confirmation in a larger, controlled, disease-specific trial, the cabazitaxel–cisplatin combination may provide a future therapeutic option for patients with advanced solid tumors. Ongoing studies are investigating the use of cabazitaxel, both as a monotherapy and combined with other agents, in a variety of solid tumors.
References
Vrignaud P, Sémiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, Riou J-F, Commercon A, Lavelle F, Bissery M-C (2013) Preclinical antitumor activity of cabazitaxel, a semi-synthetic taxane active in taxane-resistant tumors. Clin Cancer Res 19:2973–2983
Sémiond D, Sidhu SS, Bissery M-C, Vrignaud P (2013) Can taxanes provide benefit in patients with CNS tumors and in pediatric patients with tumors? An update on the preclinical development of cabazitaxel. Cancer Chemother Pharmacol 72:515–528
Siddik Z (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22:7265–7279
Vrignaud P (2011) Therapeutic synergism of cabazitaxel in combination with cisplatin in tumor-bearing mice. Cancer Res 71(Supplement 1):abstract 2522
Luo H, Yu Z, Gao H, Guan C, Xu M (2013) Efficacy and tolerability of docetaxel and cisplatin plus S-1 for advanced gastric cancer. J BUON 18:154–161
Perri F, Ionna F, Muto P, Buonerba C, Della Vittoria SG, Bosso D, Fulciniti F, Daponte A, Argenone A, Sandomenico F, Di Lorenzo G, Caponigro F (2013) Induction docetaxel-cisplatin followed by extended-field radiotherapy in patients with cervical metastases from unknown primary carcinoma. Anticancer Res 33:1135–1139
Tsuburaya A, Nagata N, Cho H, Hirabayashi N, Kobayashi M, Kojima H, Munakata Y, Fukushima R, Kameda Y, Shimoda T, Oba K, Sakamoto J (2013) Phase II trial of paclitaxel and cisplatin as neoadjuvant chemotherapy for locally advanced gastric cancer. Cancer Chemother Pharmacol 71:1309–1314
Rowinsky EK, Gilbert MR, McGuire WP, Noe DA, Grochow LB, Forastiere AA, Ettinger DS, Lubejko BG, Clark B, Sartorius SE (1991) Sequences of taxol and cisplatin: a phase I and pharmacologic study. J Clin Oncol 9:1692–1703
U.S. Department of Health and Human Services; National Institutes of Health; National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Version 4.03: June 14, 2010. Available at: http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed 20 May 2014
Mita AC, Denis LJ, Rowinsky EK, de Bono JS, Goetz AD, Ochoa L, Forouzesh B, Beeram M, Patnaik A, Molpus K, Sémiond D, Besenval M, Tolcher AW (2009) Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res 15:723–730
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Bouchet BP, Galmarini CM (2010) Cabazitaxel, a new taxane with favorable properties. Drugs Today (Barc) 46:735–742
Dizon DS, Sill MW, Gould N, Rubin SC, Yamada SD, Debernardo RL, Mannel RS, Eisenhauer EL, Duska LR, Fracasso PM (2011) Phase I feasibility study of intraperitoneal cisplatin and intravenous paclitaxel followed by intraperitoneal paclitaxel in untreated ovarian, fallopian tube, and primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol 123:182–186
Frasci G, Comella P, Parziale A, Casaretti R, Daponte A, Gravina A, De Rosa L, Gallipoli A, Comella G (1997) Cisplatin-paclitaxel weekly schedule in advanced solid tumors: a phase I study. Ann Oncol 8:291–293
Atmaca A, Al Batran SE, Werner D, Pauligk C, Guner T, Koepke A, Bernhard H, Wenzel T, Banat AG, Brueck P, Caca K, Prasnikar N, Kullmann F, Gunther DH, Koenigsmann M, Dingeldein G, Neuhaus T, Jager E (2013) A randomised multicentre phase II study with cisplatin/docetaxel vs oxaliplatin/docetaxel as first-line therapy in patients with advanced or metastatic non-small cell lung cancer. Br J Cancer 108:265–270
Ji JH, Yun T, Kim SB, Kang JH, Park JC, Cho IS, Sohn CH, Heo DS, Jang JS, Shin SW, Hwang DW, Sun JM, Park K, Ahn MJ (2012) A prospective multicentre phase II study of cisplatin and weekly docetaxel as first-line treatment for recurrent or metastatic nasopharyngeal cancer (KCSG HN07-01). Eur J Cancer 48:3198–3204
de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, Roessner M, Gupta S, Sartor AO (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376:1147–1154
Sanofi U.S. LLC. JEVTANA® (cabazitaxel) Injection, Prescribing Information. Bridgewater, NJ, USA, 2010. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/201023lbl.pdf. Accessed 20 May 2014
Masek V, Anzenbacherova E, Machova M, Brabec V, Anzenbacher P (2009) Interaction of antitumor platinum complexes with human liver microsomal cytochromes P450. Anticancer Drugs 20:305–311
Ferron GM, Dai Y, Semiond D (2013) Population pharmacokinetics of cabazitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 71:681–692
Dieras V, Lortholary A, Laurence V, Delva R, Girre V, Livartowski A, Assadourian S, Semiond D, Pierga JY (2013) Cabazitaxel in patients with advanced solid tumours: results of a phase I and pharmacokinetic study. Eur J Cancer 49:25–34
Li JL, Zhang XR, Liu JW, Chen ZY, Lin YC, Wang YD, Chen Q, Nan KJ, Song SP, Han FC, Zhu YZ, Li LY, Zheng YH, Chu DT (2006) Phase I/II clinical trial of weekly administration of docetaxel plus cisplatin for advanced non-small cell lung cancer. Zhonghua Zhong Liu Za Zhi 28:309–312
Dickgreber NJ, Fink TH, Latz JE, Hossain AM, Musib LC, Thomas M (2009) Phase I and pharmacokinetic study of pemetrexed plus cisplatin in chemonaive patients with locally advanced or metastatic malignant pleural mesothelioma or non-small cell lung cancer. Clin Cancer Res 15:382–389
Acknowledgments
The authors would like to thank participating patients and their families as well as the study co-investigators and research coordinators. Dr Wang-Gillam is a KL2 Scholar (Washington University Institute of Clinical and Translational Sciences grant KL2TR000450). Dr Sarantopoulos has received funding from the Cancer Center (Grant P30CA054174). Medical writing assistance was provided by Dr Melissa Purves of MediTech Media, funded by Sanofi. This study was funded by Sanofi.
Conflict of interest
SS, JS, MMM, JLM, AW-G, SLB, ARL and ACM have no conflicts to disclose. ACL has received research funding from Amgen, Bayer, Cephalon/TEVA, Daiichi-Sankyo, Genentech, Imclone/Lilly, Merck, Millennium, Novartis, Pfizer, Sanofi-Aventis and Zenyaku. JFD and CW are remunerated employees of Sanofi; JFD also holds stock in Sanofi. LK is employed in a remunerated consultant/advisory role by Sanofi.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lockhart, A.C., Sundaram, S., Sarantopoulos, J. et al. Phase I dose-escalation study of cabazitaxel administered in combination with cisplatin in patients with advanced solid tumors. Invest New Drugs 32, 1236–1245 (2014). https://doi.org/10.1007/s10637-014-0145-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0145-y