
Summary
Ibrutinib (formerly PCI-32765) is a specific, irreversible, and potent inhibitor of Burton’s tyrosine kinase (BTK) developed for the treatment of several forms of blood cancer. It is now an FDA-approved drug marketed under the name ImbruvicaTM (Pharmacyclics, Inc.) and successfully used as an orally administered second-line drug in the treatment of mantle cell lymphoma. Since BTK is predominantly expressed in hematopoietic cells, the sensitivity of solid tumor cells to Ibrutinib has not been analyzed. In this study, we determined the effect of Ibrutinib on breast cancer cells. We demonstrate that Ibrutinib efficiently reduces the phosphorylation of the receptor tyrosine kinases ErbB1, ErbB2 and ErbB3, thereby suppressing AKT and MAPK signaling in ErbB2-positive (ErbB2+) breast cancer cell lines. Treatment with Ibrutinib significantly reduced the viability of ErbB2+ cell lines with IC50 values at nanomolar concentrations, suggesting therapeutic potential of Ibrutinib in breast cancer. Combined treatment with Ibrutinib and the dual PI3K/mTOR inhibitor BEZ235 synergistically reduces cell viability of ErbB2+ breast cancer cells. Combination indices below 0.25 at 50 % inhibition of cell viability were determined by the Chou-Talalay method. Therefore, the combination of Ibrutinib and canonical PI3K pathway inhibitors could be a new and effective approach in the treatment of breast cancer with activated ErbB receptors. Ibrutinib could thus become a valuable component of targeted therapy in aggressive ErbB2+ breast cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common non-cutaneous cancer in Western countries, accounting for nearly one in three malignancies diagnosed in women [1]. Human epidermal growth-factor receptor 2 (ErbB2/HER2) is amplified in approximately 25 % of breast cancers and ErbB2-positive tumors represent an aggressive subtype of breast cancer [2, 3]. ErbB2 is a transmembrane tyrosine-kinase receptor belonging to the family of ErbB receptors, including ErbB1/EGFR, ErbB2/HER2, ErbB3/HER3 and ErbB4/HER4 [4, 5]. Aberrant expression of the ErbB2 receptor activates multiple downstream signaling cascades including the PI3K/AKT/mTOR pathway that is associated with poor prognosis and endocrine therapy resistance of breast cancer cells [6]. ErbB2 is the therapeutic target for the anti-ErbB2 antibody trastuzumab (Herceptin®) but resistance to Herceptin therapy is frequently observed. Therefore, new therapeutic approaches are in development for the improved treatment of ErbB2+ breast cancer including small molecular inhibitors directly targeting the tyrosine kinase activity of ErbB2 (lapatinib, neratinib, afatinib), monoclonal antibodies directed against epitopes of the extracellular domain of ErbB2 (pertuzumab) not targeted by Herceptin as well as antibody-drug conjugates (trastuzumab-DM 1) [7–9]. The majority of tyrosine kinase inhibitors (TKIs) developed thus far target the ATP-binding site of the enzyme, which is highly conserved across the human protein kinases [10]. ATP-competitive compounds inhibit the catalytic activity in a reversible manner, binding to the kinase domain of the target through weak interactions (hydrogen-bonds, van der Waals and hydrophobic interactions). In the past decade, much progress has been made in the development of a new class of potent and selective tyrosine kinase inhibitors that irreversibly inhibit their target protein via the formation of covalent bonds [11–14]. Covalently binding tyrosine kinase inhibitors carry an electrophilic functionality (warhead) that covalently interacts with a nucleophilic cysteine residue located near the ATP binding pocket [15]. As a result, the pharmacodynamic behavior of such a compound is coupled to the half-life and turnover of the target protein rather than to pharmacokinetic properties. This is an advantage for covalent inhibitors over ATP-competitive inhibitors.
Ibrutinib (formerly PCI-32765) is an orally administered, irreversible, and potent inhibitor of Bruton’s tyrosine kinase (BTK). BTK is a member of the cytoplasmic non-receptor tyrosine kinase family of TEC kinases, predominantly expressed in hematopoietic cells. BTK plays a key role in the B-cell receptor signaling pathway and is a mediator of pro-inflammatory signals [16]. Inhibition of BTK is a promising strategy for the treatment of B-cell malignancies and autoimmune disease [17, 18]. BTK belongs to a group of 11 tyrosine kinases, including the TEC family kinases, EGFR, ErbB2, ErbB4, Jak3 and BLK that contain a conserved cysteine residue adjacent to the ATP-binding site, critical for covalent inhibition by tyrosine kinase inhibitors [14, 19]. Ibrutinib binds covalently to the cysteine-481 residue at the active site of BTK, resulting in potent inhibition of kinase activity, with an IC50 of 0.5 nM for more than 24 h [20]. Since November 2013, Ibrutinib is approved by the FDA, marketed under the name Imbruvica (Pharmacyclics Inc.) and is successfully used for second line treatment of mantle cell lymphoma [21]. In this study, we performed an initial analysis of the effect of Ibrutinib on breast cancer cells. We found that Ibrutinib is an effective inhibitor of ErbB receptor phosphorylation and of the viability of ErbB2+ breast cancer cells, suggesting therapeutic use of Ibrutinib in breast cancer.
Materials and methods
Cell culture and reagents
The human breast cancer cell lines MCF-7, T-47D, HCC-1954, SKBR-3, BT-474, SUM-159PT and MDA-MB 231 were kindly provided by Peter K. Vogt and cultured by standard conditions as described and recommended by ATCC. MDA-MB 468 and BT-20 cells were kindly provided by Klaus Pantel (Institute of Tumor Biology, University Medical Center Hamburg Eppendorf, Germany) and cultured by standard conditions as described and recommended by ATCC. Murine Balb-neuT-derived ErbB2-positive breast cancer cell line C4 was established, characterized and cultured as described previously [22]. All media and supplements were purchased from Invitrogen (USA). All inhibitors were supplied by Selleckchem (USA) and have been diluted in DMSO (Sigma, USA) as recommended.
Cell viability assay
For the alamar blue cell viability assay, 5 × 103 cells were seeded per 96 well and allowed to adhere overnight followed by treatment with the corresponding inhibitor for the indicated time and concentrations or DMSO as a control. Prior to measurement, the medium was removed, and 100 μl medium containing 5 ng/ml Resazurin (Sigma, USA) was added to each well. Cells were incubated for 60–240 min at 37 °C in a humified atmosphere. Fluorescence based absorption was measured at 540 nm on a micro plate reader (Tekan, Switzerland).
Western blot analysis and immunoprecipitation
For Western blot analysis and immunoprecipitation (IP), protein lysates were prepared by solubilizing cells in cell lysis buffer (Cell Signaling, USA). Proteins were resolved by SDS-polyacrylamide gel electrophoresis and were transferred to nitrocellulose paper (GE Healthcare, USA). Membranes were probed with the following primary antibodies: pSTAT3(Y705), STAT3, pAKT(S473), AKT, EGFR, pEGFR(Y1068), ErbB2, pErbB2(Y877), ErbB3, pErbB3(Y1289), pGSK3β(S9), pMAPK and GAPDH (Cell Signaling, USA), phospho-tyrosine, MAPK, HSC70 (Santa Cruz, USA) and BMX (Abgent, USA) followed by probing with secondary antibodies (GE Healthcare, USA). Western blots were developed using standard procedures. Direct immunoprecipitation was carried out by coupling of 2.5 μg of the corresponding antibody for 2 h, rotating at 4 °C to 40 μl of equilibrated 50:50 slurry of protein G-Sepharose beads (GE Healthcare, USA). Beads were washed twice in cell lysis buffer before 100 μg of protein from whole cell lysates in a total volume of 500 μl was added. Proteins were precipitated overnight, rotating at 4 °C. Beads were washed four times in cell lysis buffer. Sample loading buffer was added to beads and probes were heated 5 min at 100 °C. Whole samples were analyzed by western blot technique probed with the indicated antibodies. Subsequently, nitrocellulose membrane was incubated with secondary antibody (GE Healthcare, USA), either anti-rabbit or anti-mouse, dependent on the antibody used in the IP to detect IgG levels for sample correction.
Statistical analysis
Drug interactions were analyzed based on the median effect method of Chou and Talalay [23]. CalcuSyn software (Biosoft, Cambridge, UK) was used to calculate the combination index (CI) as described previously [24]. IC50 values were calculated using GraphPad Prism 4.0 software.
Results
Ibrutinib potently inhibits cell viability of ErbB2+ breast cancer cells
To analyze the effect of Ibrutinib on breast cancer cell viability, ten breast cancer cell lines of different clinical subtypes i.e. ER+, ErbB2+ and triple-negative (TN), were used (Table 1). Cell viability of breast cancer cells was measured by the alamar blue assay after 72 h of treatment with Ibrutinib at the indicated concentrations. As shown in Fig. 1a, the viability of ER + (MCF-7 and T-47D) and TN (SUM-159PT, MDA-MB 231, MDA-MB 468 and BT-20) breast cancer cells was not significantly attenuated after treatment with Ibrutinib at increasing concentrations. Even at the highest applied concentration of 6.4 μM of Ibrutinib, 50 % inhibition of cell viability was not observed. In contrast, ErbB2+ (BT-474, SKBR-3, HCC-1954 and C4) breast cancer cell lines were highly sensitive to Ibrutinib treatment (Fig. 1a). While HCC-1954 cells were slightly more resistant (IC50 = 180 nM), 50 % reduction of cell viability of BT-474, SKBR-3 and C4 cells was reached at doses below 5 nM (Fig. 1b and c). These data show that Ibrutinib specifically and potently inhibits cell viability of ErbB2+ breast cancer cells. IC50 values of 2.1, 2.0 and 4.3 nM were calculated for the ErbB2+ breast cancer cell lines BT-474, SKBR-3, and C4, respectively (Fig. 1c and Table 1).

Ibrutinib potently inhibits cell viability of ErbB2+ breast cancer cells. Cell viability of breast cancer cell lines was measured after 72 h of treatment with Ibrutinib at the indicated concentrations by alamar blue assay. Shown are mean values (n ≥ 3), +/− SD
AKT and MAPK phosphorylation is reduced after treatment with Ibrutinib in ErbB2+ breast cancer cells
In order to determine the mechanism of inhibition of cell viability of ErbB2+ breast cancer cells by Ibrutinib, common cancer related signaling pathways, including MAPK, AKT and STAT3, were analyzed by Western blot after treatment with Ibrutinib (10 μM for 24 h). Ibrutinib reduced phosphorylation of MAPK in all investigated cell lines with the exception of the kras and braf mutated cell line MDA-MB 231, whereas phosphorylation of AKT was significantly reduced only in ErbB2+ breast cancer cell lines (Fig. 2a). In comparison, the selective PI3K inhibitor GDC-0941 reduced PI3K-mediated AKT phosphorylation in all investigated cell lines, indicating that Ibrutinib specifically inhibits AKT phosphorylation of ErbB2+ cell lines (Fig. 2a). There was no correlation between sensitivity to Ibrutinib and phosphorylation status of STAT3 (Fig. 2a). To further analyze the effect of Ibrutinib on phosphorylation of AKT, lower doses of Ibrutinib were tested and applied for 1 h to different breast cancer cell lines (Fig. 2b). As shown in Fig. 2b, treatment with Ibrutinib resulted in a concentration-dependent decrease of AKT phosphorylation in ErbB2+ cell lines BT-474 and C4 (Fig. 2b). Similar results were obtained for the ErbB2+ breast cancer cell lines SKBR-3 and HCC-1954 (Online Resource 1). In addition, phosphorylation of the AKT substrate GSK3β at serine residue 9 decreased after treatment with Ibrutinib in a dose-dependent manner, indicating that downstream signaling events of AKT are also inhibited (Fig. 2b). In contrast, Ibrutinib-resistant cell lines MCF-7 and BT-20 showed less or no attenuation of AKT phosphorylation after treatment with Ibrutinib (Fig. 2b). Similar results were observed for other Ibrutinib resistant breast cancer cell lines (data not shown). The phosphorylation of STAT3 at Y705 was not affected by a 1-h treatment with Ibrutinib (Online Resource 1). These data indicate that Ibrutinib inhibits ErbB2-mediated activation of AKT in ErbB2+ breast cancer cells but has no detectable effect on the phosphorylation of STAT3.

AKT and MAPK phosphorylation is reduced after treatment with Ibrutinib in ErbB2+ breast cancer cells. Breast cancer cells have been treated with the PI3K inhibitor GDC-0941 or Ibrutinib at the indicated concentrations for a 24 h or b 1 h. Cells have been lysed, subjected to western blot analysis and probed with the indicated antibodies
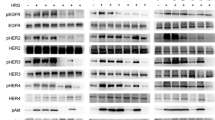
Phosphorylation of ErbB1, 2 and 3 is reduced after treatment with Ibrutinib in ErbB2+ breast cancer cells
We monitored the activity of ErbB2 in breast cancer cells by determining the level of phosphorylation of this receptor tyrosine kinase after treatment with Ibrutinib and the PI3K inhibitor GDC-0941 in ErbB2-positive HCC-1954 and SKBR-3 cells and in ErbB2-negative MCF-7 cells (Fig. 3a). Treatment with Ibrutinib resulted in reduced phosphorylation of ErbB2 in HCC-1954 and SKBR-3 cells which show constitutive activation of ErbB2, whereas phosphorylated ErbB2 was not detectable in MCF7 cells (Fig. 3a). No significant reduction of phosphorylation of ErbB2 was observed after treatment with the PI3K inhibitor GDC-0941 (Fig. 3a). In ErbB2+ cells the phosphorylation of ErbB2 was almost completely abrogated at a concentration of 0.1 μM Ibrutinib, whereas the expression of ErbB2 was not affected (Fig. 3b). For ErbB2-positive breast cancer, hetero-dimerization of ErbB2 with its family member ErbB3 is an essential event [25]. Therefore we also determined the phosphorylation of ErbB3 after treatment with Ibrutinib (Fig. 3b). In accordance with the observations on ErbB2, ErbB3 phosphorylation was completely abolished after treatment with Ibrutinib at a concentration of 0.1 μM. The phosphorylation of all three members of the EGFR family, ErbB1, ErbB2 and ErbB3 was reduced by Ibrutinib in a dose-dependent manner in all investigated cell lines that showed activation of these receptors (Fig. 3c). These results demonstrate that Ibrutinib is a potent inhibitor of ErbB1, 2 and 3 phosphorylation and downstream signaling at clinically achievable concentrations of this compound.

Phosphorylation of ErbB1, 2 and 3 is efficiently reduced after treatment with Ibrutinib in ErbB2+ breast cancer cells. Breast cancer cells have been treated with the PI3K inhibitor GDC-0941 or Ibrutinib at the indicated concentrations for a, b 24 h or c 1 h. Cells have been lysed, subjected to immunoprecipitation with the indicated antibodies followed by western blot analysis a and b or c directly subjected to western blot analysis and probed with the indicated antibodies
Involvement of the TEC kinase family member BMX in the reduction of ErbB receptor phosphorylation after treatment with Ibrutinib in ErbB2+ breast cancer cells
The primary target of Ibrutinib is Burton’s tyrosine kinase, a member of the TEC tyrosine kinase family. The TEC protein family includes the ubiquitously expressed BMX kinase which can be inhibited by Ibrutinib [26]. In order to test whether the effect of Ibrutinib on ErbB proteins is mediated by an inhibition of BMX, we analyzed the expression and phosphorylation of this kinase after treatment with Ibrutinib (Fig. 4a, b). Ibrutinib inhibited the phosphorylation of BMX but this activity required a drug concentration of 1 μM, about ten times the dose needed for inhibiting the phosphorylation of the ErbB proteins. We conclude that the inhibition of ErbB phosphorylation by Ibrutinib is not a consequence of reduced BMX phosphorylation. BMX physically interacts with ErbB3 [27]; it is also able to phosphorylate receptor tyrosine kinases, including the insulin receptor, resulting in full auto-phosphorylation activity of these receptors [28]. In order to further analyze the impact of BMX on the mechanism of inhibition of cell viability of ErbB2+ breast cancer cells by Ibrutinib, the interaction of BMX with ErbB receptors was analyzed by co- immunoprecipitation. As shown in Fig. 4c, BMX is co-precipitated with ErbB3 in MCF-7 cells. In contrast, only smaller amounts of BMX were co-precipitated with ErbB3 in ErbB2+ cell lines (Fig. 4c). Immunoprecipitation of ErbB2 followed by Western blot analysis with BMX antibody suggests that there is no interaction between ErbB2 and BMX (Fig. 4d). These results suggest that Ibrutinib inhibits ErbB receptor phosphorylation without the involvement of inhibition of the TEC kinase BMX.

Involvement of the TEC kinase family member BMX in the reduction of ErbB receptor phosphorylation after treatment with Ibrutinib. a. Breast cancer cells have been lysed and subjected to western blot analysis probed with the indicated antibodies. Phosphorylated proteins were immunoprecipitated from whole cell lysates (WCL) with phospho-tyrosine (pY) antibody followed by western blot analysis with BMX antibody to detect phosphorylated BMX. b. SKBR-3 cells were treated with Ibrutinib at the indicated concentrations, lysed, subjected to immunoprecipitation with pY antibody followed by western blot analysis with BMX antibody. c. and d. Breast cancer cells were lysed, subjected to immunoprecipitation with the indicated antibodies followed by western blot analysis probed with the indicated antibodies (1stSN: first supernatant after immunoprecipitation)
A combination of Ibrutinib and the dual PI3K/mTOR inhibitor BEZ235 synergistically inhibits cell viability of ErbB2+ breast cancer cells
Since inhibition of PI3K/AKT signaling seems to be a key mechanism by which Ibrutinib inhibits viability of ErbB2+ breast cancer cells, we analyzed whether the effect of Ibrutinib can be increased by combining Ibrutinib with canonical PI3K pathway inhibitors. We therefore combined Ibrutinib with the AKT inhibitor MK-2206, the mTOR inhibitor Rapamycin, the PI3K inhibitor GDC-0941 or the dual PI3K/mTOR inhibitor BEZ235. The combination of Ibrutinib and BEZ235 was highly synergistic in all investigated ErbB2+ breast cancer cells (Fig. 5a). Combination indices were calculated by the Chou-Talalay method for drug combination for HCC-1954, SKBR-3 and BT-474 cells of 0.22, 0.14 and 0.001, respectively at 50 % inhibition of cell viability (Fig. 5b). All other drug combinations had no significant synergistic effect on cell viability except for Ibrutinib and GDC-0941 which synergistically affected a single cell line, HCC-1954 (Chou-Talalay combination index of 0.57 at 50 % inhibition of cell viability, data not shown). Our data suggest that a combination of Ibrutinib with canonical PI3K pathway inhibitors like BEZ235 offers superior cytocidal potency. These results could form the basis for the development of new treatment strategies for ErbB2+ breast cancer.

Combination of Ibrutinib and the dual PI3K/mTOR inhibitor BEZ235 synergistically inhibits cell viability of ErbB2+ breast cancer cells. A. Cell viability of ErbB2+ breast cancer cells was measured by alamar blue assay after 72 h of treatment with Ibrutinib, BEZ235 or in combination with both as indicated. Shown are mean values (n ≥ 3), +/− SD. B. The combination indices (CI) were calculated by the Chou-Talalay method for drug combination
Discussion
ErbB2+ breast cancer occurs in about 25 % of human breast cancer and is associated with a high metastatic potential and poor prognosis. New targeted therapeutic approaches for endocrine and herceptine® resistant ErbB2+ breast cancer patients are urgently needed. Here, we demonstrate that the FDA-approved BTK inhibitor Ibrutinib (ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and cell viability of ErbB2+ breast cancer cells. For covalent inhibition of tyrosine kinases (TKs) a cysteine residue located in close proximity to the ATP binding pocket is critical [15, 29]. Among 100 TKs, encoded in the human genome, 11 share this cysteine residue. These kinases include BTK, BMX, EGFR, ErbB2 and ErbB4 [19, 30]. In vitro kinase activity assays using Ibrutinib revealed IC50 values of 171 nM and 200 nM for inhibition of kinase activity of EGFR and ErbB2, respectively [26]. However, our data demonstrate that Ibrutinib is a highly effective and specific inhibitor of ErbB1, ErbB2 and ErbB3 phosphorylation with IC50 values for inhibition of cell viability in the lower nanomolar range of all ErbB2+ cell lines investigated. This high sensitivity of ErbB2+ breast cancer cells on cell viability might be explained by an addiction of these cells to survive and proliferate via ErbB2/PI3K/AKT/mTOR signaling, as indicated by our results.
Moreover, we have shown that the combination of Ibrutinib with the dual PI3K/mTOR inhibitor BEZ235 synergistically inhibits cell viability of ErbB2+ breast cancer cells. Recently, a synergistic interaction between Ibrutinib and a wide range of compounds targeting the PI3K/AKT/mTOR-signaling cascade, including BEZ235, was demonstrated in an activated B-cell-like subtype of diffuse large B-cell lymphoma [31]. The development of efficient drug combination regimes in the treatment of cancer is a promising therapeutic strategy. Collectively, our findings suggest a potential use of Ibrutinib (ImbruvicaTM) in the treatment of ErbB2+ breast cancer. Furthermore, the therapeutic potential of Ibrutinib in breast cancer therapy might be increased by the combination of Ibrutinib with canonical PI3K pathway inhibitors like BEZ235. Future studies need to address a potential use of Ibrutinib in the treatment of different tumor entities where activation of ErbB receptors is frequently observed, i.e. lung and colorectal cancer.
References
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F (2013) Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 49(6):1374–1403. doi:10.1016/j.ejca.2012.12.027
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235(4785):177–182
Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM (2003) Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol 200(3):290–297. doi:10.1002/path.1370
Arteaga CL, Engelman JA (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25(3):282–303. doi:10.1016/j.ccr.2014.02.025
Coussens L, Yang-Feng TL, Liao YC, Chen E, Gray A, McGrath J, Seeburg PH, Libermann TA, Schlessinger J, Francke U et al (1985) Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230(4730):1132–1139
Arpino G, Wiechmann L, Osborne CK, Schiff R (2008) Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev 29(2):217–233. doi:10.1210/er.2006-0045
Modjtahedi H, Cho BC, Michel MC, Solca F (2014) A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn Schmiedeberg’s Arch Pharmacol. doi:10.1007/s00210-014-0967-3
Rexer BN, Arteaga CL (2012) Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog 17(1):1–16
Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462(7276):1070–1074. doi:10.1038/nature08622
Tibes R, Trent J, Kurzrock R (2005) Tyrosine kinase inhibitors and the dawn of molecular cancer therapeutics. Annu Rev Pharmacol Toxicol 45:357–384. doi:10.1146/annurev.pharmtox.45.120403.100124
Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A (2013) Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 3(12):1404–1415. doi:10.1158/2159-8290.CD-13-0314
Garske AL, Peters U, Cortesi AT, Perez JL, Shokat KM (2011) Chemical genetic strategy for targeting protein kinases based on covalent complementarity. Proc Natl Acad Sci U S A 108(37):15046–15052. doi:10.1073/pnas.1111239108
Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM (2007) Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol 3(4):229–238. doi:10.1038/nchembio866
Cohen MS, Zhang C, Shokat KM, Taunton J (2005) Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science 308(5726):1318–1321. doi:10.1126/science1108367
Zhang J, Yang PL, Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9(1):28–39. doi:10.1038/nrc2559
Lopez-Herrera G, Vargas-Hernandez A, Gonzalez-Serrano ME, Berron-Ruiz L, Rodriguez-Alba JC, Espinosa-Rosales F, Santos-Argumedo L (2014) Bruton’s tyrosine kinase—an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol 95(2):243–250. doi:10.1189/jlb.0513307
Hendriks RW, Yuvaraj S, Kil LP (2014) Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer 14(4):219–232. doi:10.1038/nrc3702
Buggy JJ, Elias L (2012) Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol 31(2):119–132. doi:10.3109/08830185.2012.664797
Leproult E, Barluenga S, Moras D, Wurtz JM, Winssinger N (2011) Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem 54(5):1347–1355. doi:10.1021/jm101396q
Brown JR (2013) Ibrutinib (PCI-32765), the first BTK (Bruton’s tyrosine kinase) inhibitor in clinical trials. Curr Hematol Malignancy Rep 8(1):1–6. doi:10.1007/s11899-012-0147-9
Dias AL, Jain D (2014) Ibrutinib: a New frontier in the treatment of chronic lymphocytic leukemia by Bruton’s tyrosine kinase inhibition. Cardiovasc Hematol Agents Med Chem 11(4):265–271
Grabinski N, Mollmann K, Milde-Langosch K, Muller V, Schumacher U, Brandt B, Pantel K, Jucker M (2014) AKT3 regulates ErbB2, ErbB3 and estrogen receptor alpha expression and contributes to endocrine therapy resistance of ErbB2(+) breast tumor cells from Balb-neuT mice. Cell Signal 26(5):1021–1029. doi:10.1016/j.cellsig.2014.01.018
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B, Jucker M (2012) Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer 11:85. doi:10.1186/1476-4598-11-85
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A 100(15):8933–8938. doi:10.1073/pnas.1537685100
Wu H, Wang W, Liu F, Weisberg EL, Tian B, Chen Y, Li B, Wang A, Wang B, Zhao Z, McMillin DW, Hu C, Li H, Wang J, Liang Y, Buhrlage SJ, Liang J, Liu J, Yang G, Brown JR, Treon SP, Mitsiades CS, Griffin JD, Liu Q, Gray NS (2014) Discovery of a potent covalent BTK inhibitor for B-cell lymphoma. ACS Chem Biol. doi:10.1021/cb4008524
Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP (2007) Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J Biol Chem 282(45):32689–32698. doi:10.1074/jbc.M703412200
Chen S, Jiang X, Gewinner CA, Asara JM, Simon NI, Cai C, Cantley LC, Balk SP (2013) Tyrosine kinase BMX phosphorylates phosphotyrosine-primed motif mediating the activation of multiple receptor tyrosine kinases. Sci Signal 6(277):ra40. doi:10.1126/scisignal.2003936
Carmi C, Mor M, Petronini PG, Alfieri RR (2012) Clinical perspectives for irreversible tyrosine kinase inhibitors in cancer. Biochem Pharmacol 84(11):1388–1399. doi:10.1016/j.bcp.2012.07.031
Hur W, Velentza A, Kim S, Flatauer L, Jiang X, Valente D, Mason DE, Suzuki M, Larson B, Zhang J, Zagorska A, Didonato M, Nagle A, Warmuth M, Balk SP, Peters EC, Gray NS (2008) Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg Med Chem Lett 18(22):5916–5919. doi:10.1016/j.bmcl.2008.07.062
Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, Goldlust IS, Yasgar A, McKnight C, Boxer MB, Duveau DY, Jiang JK, Michael S, Mierzwa T, Huang W, Walsh MJ, Mott BT, Patel P, Leister W, Maloney DJ, Leclair CA, Rai G, Jadhav A, Peyser BD, Austin CP, Martin SE, Simeonov A, Ferrer M, Staudt LM, Thomas CJ (2014) High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 111(6):2349–2354. doi:10.1073/pnas.1311846111
Acknowledgments
We thank Peter K. Vogt for guidance and support during these studies and for his help in the preparation of the manuscript. We thank Mailo Timm for perfect technical assistance.
Financial support
N.G. was funded by the Deutsche Krebshilfe (Dr. Mildred Scheel scholarship). This work was further supported by the National Cancer Institute under award number R01 CA078230 to Peter K. Vogt. This is manuscript 27046 of The Scripps Research Institute.
Conflict of interest
The authors disclose no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 209 kb)
Rights and permissions
About this article

Cite this article
Grabinski, N., Ewald, F. Ibrutinib (ImbruvicaTM) potently inhibits ErbB receptor phosphorylation and cell viability of ErbB2-positive breast cancer cells. Invest New Drugs 32, 1096–1104 (2014). https://doi.org/10.1007/s10637-014-0141-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0141-2