
Summary
Background This phase 1b study evaluated an enteric-coated tablet (ECT) formulation of the investigational Aurora A kinase inhibitor, alisertib (MLN8237). Methods Patients with advanced, non-hematologic malignancies received oral alisertib ECT for 7 d BID followed by 14 d treatment-free (21-day cycles; 3 + 3 dose escalation schema). Objectives were to assess safety, pharmacokinetics, and antitumor activity, and to define a recommended phase 2 dose (RP2D) of alisertib. Results 24 patients were treated. Median age was 57 years. Patients received a median of 2 cycles (range 1–12). The RP2D was determined as 50 mg BID for 7 d (21-day cycles). A cycle 1 dose-limiting toxicity of grade 4 febrile neutropenia was observed in 1 of 13 patients at RP2D. The most common drug-related adverse event (AE) was neutropenia (50 %). At doses ≥40 mg BID, 7 patients had drug-related AEs that were serious but largely reversible/manageable by dose reduction and supportive care, including 3 with febrile neutropenia. Pharmacokinetic data were available in 24 patients. Following administration of alisertib ECT, the plasma peak concentration of alisertib was achieved at ~3 h; systemic exposure increased with increasing dose over 10–60 mg BID. Mean t½ was ~21 h following multiple dosing. Renal clearance was negligible. Nine patients achieved stable disease (3.98*, 5.59, 1.28*, 2.56, 5.45*, 3.48, 3.15, 8.31, and 6.93* months; *censored). Conclusions Alisertib ECT was generally well tolerated in adults with advanced, non-hematologic malignancies. The RP2D is 50 mg BID for 7 d and is being evaluated in ongoing phase 2 studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Aurora kinases (A, B, and C) belong to a family of oncogenic serine/threonine kinases that play a key role in proper mitotic execution [1]. Aurora A kinase (AAK), the most well-characterized member of the family, is essential for centrosome function and maturation, spindle assembly, chromosome segregation, and mitotic entry [2–4]. Inhibition of AAK by specific small molecule inhibitors or knockdown of AAK by small RNA interference causes abnormal spindle formation, mitotic defects, senescence, and cell death [4–6]. AAK has been implicated in oncogenesis [7] and correlates with more histologically aggressive forms of cancer [8, 9]. Indeed, over-expression or amplification of AAK and/or antitumor activity of AAK inhibitors have been observed in a variety of tumor types including head and neck cancer [10], non-small cell lung cancer (NSCLC) [11], small cell lung cancer (SCLC) [12], breast cancer [13], upper gastrointestinal cancer (including lower esophageal, gastroesophageal junction, gastric cardia, and distal gastric) [14], and esophageal cancer [15, 16], suggesting that these tumor types may be likely to respond to AAK inhibition as a clinical therapeutic strategy.
Alisertib (MLN8237) is an investigational, oral, and selective inhibitor of AAK [17]. In preclinical studies using xenograft models, alisertib demonstrated potent AAK inhibition and high antitumor activity in a wide range of tumor cell types [8, 17–19]. Subsequent phase 1 studies have shown that the pharmacodynamic effects of alisertib are consistent with AAK inhibition with 7-day exposure to alisertib resulting in abnormal spindle formation and chromosomal aberrations [19, 20].
Recent clinical studies in patients with solid tumors have shown that up to 3 years of cyclic alisertib was feasible in patients who experienced objective responses or prolonged disease control and that repeat cycles were generally tolerable given conventional outpatient monitoring [21–23]. These early clinical studies used a buffered powder-in-capsule (PIC) formulation of alisertib on the basis of preclinical evaluations which identified a reduced solubility in acidic solution. An enteric-coated tablet (ECT) formulation was subsequently developed to bypass stomach acid and support absorption in the small intestine. In a relative bioavailability analysis of phase 1 data, the ECT formulation was shown to have generally similar systemic exposures to the PIC formulation [24] and is the preferred formulation for future clinical development. Here we report data from a phase 1b study of alisertib ECT, which assessed the safety, pharmacokinetics (PK), and antitumor activity in adults with non-hematologic malignancies (ClinicalTrials.gov identifier: NCT01045421).
Patients and methods
This open-label phase 1b study of the ECT formulation of alisertib in patients with non-hematologic malignancies was conducted at two centers: the University of Texas M.D. Anderson Cancer Center and the University of Utah Huntsman Cancer Institute. The study was carried out in accordance with the principles stated in the Declaration of Helsinki and Good Clinical Practice. Institutional Review Boards provided approval of the study, and all patients provided written informed consent. The primary phase 1 objective was to assess the safety and tolerability of alisertib ECT in a 7-day dosing schedule and to define a recommended phase 2 dose (RP2D). The secondary objectives were to characterize the PK of alisertib ECT administered in a seven-day schedule and to describe antitumor activity.
Patients
Patients with advanced, unresectable, non-hematologic malignancy were eligible for inclusion. Study participants were required to be at least 18 years of age and have an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 or 1, as well as adequate hematologic, renal, and hepatic function, defined as: total bilirubin ≤1.5 × upper limit of normal (ULN); aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤3 × ULN (elevations ≤5 × ULN permitted if reasonably ascribed to underlying cancer or liver metastases); creatinine clearance ≥30 mL/min (Cockcroft-Gault); absolute neutrophil count (ANC) ≥1,500/mm3 without growth factor support; platelet count ≥75,000/mm3 without transfusion requirement. Exclusion criteria included: an inability to swallow oral medication; receipt of more than two previous cytotoxic regimens (four for breast cancer) in the setting of metastatic or recurrent disease; prior treatment with AAK targeted agents (including alisertib); systemic anticancer treatment within 21 d preceding the first dose of alisertib; prior high-dose chemotherapy, allogeneic bone marrow or other organ transplantation, or radiotherapy to ≥25 % of the bone marrow; symptomatic brain metastases; and diagnosis of another malignancy within 2 years before the first dose of alisertib.
Study design
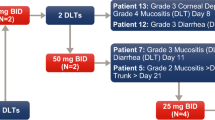
Patients received oral alisertib (provided by Takeda Pharmaceuticals International Co.) ECT for 7 d followed by a 14-day treatment-free period in 21-day cycles. Doses were escalated across 5 cohorts (10, 20, 40, 50, and 60 mg twice a day [BID]) following a standard 3 + 3 dose escalation design until the maximum tolerated dose (MTD) was reached, at which point the MTD dose level was expanded to 12 patients (the starting dose of alisertib in the initial patient cohort was taken with 240 mL water after a 2-hour fast). An additional cohort at the MTD of up to 12 patients with pancreatic cancer was also planned. Initially, 1 patient was enrolled to the first cohort and, in the absence of grade 2 or higher toxicity, the second cohort was opened with 3 patients. The MTD was defined as the highest dose at which dose-limiting toxicity (DLT) occurred in less than 2 of 6 patients during the first cycle. DLTs were defined according to the National Cancer Institute - Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0 [25] as any of the following events considered by the investigator to be related to alisertib: grade 4 neutropenia lasting longer than 7 consecutive days or with coincident fever; grade 4 thrombocytopenia lasting longer than 7 d, grade 3 thrombocytopenia with clinically significant bleeding, or platelet count less than 10,000/mm3 at any time; a delay of more than 7 d in the initiation of a subsequent cycle due to treatment-related toxicity; any grade 3 or higher non-hematologic toxicity, except grade 3 or higher nausea/emesis in the absence of optimal anti-emetic therapy, grade 3 or higher diarrhea in the absence of loperamide, grade 3 or higher fatigue lasting less than 1 week; or other grade 3 or higher non-hematologic toxicity that could be controlled to grade 2 or less with appropriate treatment. Treatment was continued until disease progression or unacceptable treatment-related toxicity. Use of myeloid growth factors was permitted from screening according to the ASCO 2006 guidelines.
Assessments
Adverse events (AEs) and serious AEs (SAEs) were graded according to NCI-CTCAE version 4.0 in all patients who received at least one dose of study drug. Safety was assessed with clinical and laboratory evaluation, ECGs, and vital sign measurements. Clinical tumor responses were based on investigator assessments taken every 6 weeks according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. [26] The response-evaluable population included all patients with measurable disease who received at least 1 dose of alisertib and had at least 1 response assessment.
Pharmacokinetics
PK parameters were calculated by non-compartmental analysis using a validated liquid chromatography tandem mass spectrometry (LC-MS/MS) method.
In cycle 1, blood samples (3 mL) for PK analyses were drawn within 1 hour prior to the morning dose of alisertib, and at 0.5, 1, 2, 3, 4, 6, 8, and 12 h post-dose on days 1 and 7. Additional samples were collected in the morning of days 8, 9, 10, 11, and 13 at 12, 36, 60, 84, and 132 h, respectively, after the day 7 afternoon dose of alisertib. In cycle 2, a single blood sample for PK analysis was collected on day 8 to contribute to alisertib population PK analysis, which will be reported separately.
Urine samples for PK analysis were collected from patients who were treated with the 40 mg BID dose level or higher. The urine PK time points included pre-dose on day 1 and during the 0–12 h post-dose interval following the morning dose on day 7.
Results
Patients
A total of 24 patients were treated, at doses of 10 (n = 1), 20 (n = 3), 40 (n = 4), 50 (n = 13), or 60 mg (n = 3) BID. Patient demographics are shown in Table 1. The data cut off for this analysis was December 2011. The median age of patients was 57 years (range 34–81), and 63 % were male. Although an expansion cohort of up to 12 pancreatic cancer patients was planned, only 1 patient was recruited who met eligibility requirements, leading to closure of the cohort due to inadequate enrollment, and this patient was analyzed as part of the 50 mg BID dose group.
Safety
Overall, 20 (83 %) patients experienced drug-related AEs (Table 2), with the most frequent including neutropenia (50 %), alopecia, diarrhea, leukopenia, nausea, and stomatitis (42 % each). Ten (42 %) patients had no drug-related AEs greater than grade 2. Dose reductions were necessary in 5 (21 %) patients for 10 AEs, including neutropenia (3 patients, 13 %), thrombocytopenia (2 patients, 8 %), and febrile neutropenia, diarrhea, vomiting, and lymphopenia (each, 1 patient, 4 %). Irrespective of causal relationship, 7 (29 %) patients discontinued treatment due to AEs (alisertib dose: 20 mg [n = 1], 50 mg [n = 4], 60 mg [n = 2]), including confusional state (drug-related), spinal disorder, spinal cord compression, urinary retention, febrile neutropenia (drug-related), fatigue, hyperbilirubinemia, hyponatremia, hypophosphatemia, and progression of pancreatic carcinoma; 1 patient discontinued due to a combination of anemia, gastrointestinal hemorrhage, lymphocytopenia, and leukocytopenia.
At 60 mg BID, alisertib-related grade 4 neutropenia occurred in all 3 (100 %) patients, and 2 of these 3 patients experienced treatment-related SAEs, including febrile neutropenia, neutropenia, diarrhea, abdominal pain, anemia, pyrexia, and pneumonia. The 60 mg BID dose level was poorly tolerated, and was therefore considered to be above the MTD. At the 50 mg BID dose level, 1 of 6 patients experienced a DLT of grade 4 febrile neutropenia. The MTD and RP2D were determined to be 50 mg BID × 7 d in a 21-day cycle.
At the MTD of 50 mg BID, 4 of 13 (31 %) patients had no drug-related AEs greater than grade 2. The most frequent grade 3 or higher drug-related AEs were neutropenia (n = 7) and leukopenia (n = 6). Across all dose groups, drug-related grade 3 or higher hematologic and gastrointestinal AEs were reported in 50 % and 25 % of patients, respectively, and events were generally manageable. Eight (33 %) patients reported any grade of drug-related central nervous system (CNS) AEs, including hypersomnia (n = 5), memory impairment (n = 3), and dizziness (n = 2). Treatment-related fatigue was observed in 8 (33 %) patients.
Seven (29 %) patients had drug-related SAEs (all at doses of 40 mg BID or higher), of whom 4 were in the MTD cohort. In addition to those experienced at 60 mg BID (detailed above), other SAEs included: neutropenia, leukopenia, vomiting (each n = 2), febrile neutropenia, nausea, stomatitis, and failure to thrive (each n = 1). At the 50 mg starting dose, 2 patients with gastrointestinal malignancies died during the first treatment cycle, but were not considered to be related to alisertib. These included a patient with pancreatic cancer who died due to disease progression, and a patient with colorectal cancer who enrolled with poor prognostic features including stage IV disease metastatic to multiple sites (lung, liver, lymph nodes), coupled with elevated lactate dehydrogenase and reduced serum albumin, who died due to acute respiratory distress related to the colorectal cancer and metastases to the lung.
PKs of alisertib following ECT dosing
Twenty-four patients completed the protocol-specified dosing and PK assessments for evaluability. Following oral administration, alisertib was absorbed with an overall median Tmax of 3 h (range 0–11). Steady-state (day 7) systemic exposures increased with increasing doses over the 10–60 mg BID dose range (Table 3). The overall mean terminal half-life following multiple dose administration was approximately 21 h, the overall mean peak/trough ratio was 2.2, and the accumulation ratio was 2.4 following BID dosing. Mean plasma-concentration time profiles of alisertib following single (day 1) and multiple dose (day 7) are shown in Fig. 1. Geometric mean steady-state trough concentrations at the RP2D of 50 mg BID exceeded concentrations associated with saturating levels of pharmacodynamic (PD) activity in murine xenograft studies (1 μM) [17].

Mean concentration-time profiles of alisertib administered as ECT following a single (day 1) and b multiple (day 7) oral administration
Urine was collected over 0 to 12 h postdose on day 7 to permit assessment of alisertib renal clearance in 12 patients (10 patients received 50 mg BID and 2 patients received 60 mg BID). The renal clearance of alisertib in humans was less than 0.1 % of the apparent oral clearance.
Antitumor efficacy
Patients received a median of 2 cycles (range 1–12). Sixteen (67 %) patients were response-evaluable. The remaining eight (33 %) patients discontinued alisertib before the first post-baseline response assessment because of toxicity (n = 4) or patient withdrawal (n = 4). No responses by RECIST were observed. Nine patients had stable disease (3.98*, 5.59, 1.28*, 2.56, 5.45*, 3.48, 3.15, 8.31, and 6.93* months; *censored observations), including 3 patients who received at least 6 cycles (1 patient with ovarian cancer and 1 patient with colorectal cancer, each of whom received 8 cycles, and 1 patient with neuroendocrine carcinoma of the pancreas who received 12 cycles), with non-progressive disease durable for up to 8 months. Criteria for completion of treatment were disease progression in 11 (46 %) patients and symptomatic deterioration in 5 (21 %); global deterioration of health status requiring discontinuation of treatment without objective evidence of disease progression). Four (17 %) patients discontinued due to AEs.
Tumor reduction less than 50 % of longest diameter, which did not meet RECIST criteria for partial response was observed in a patient with neuroendocrine carcinoma of the pancreas, which was metastatic to the liver, who received 12 cycles in total (Fig. 2). In this patient, the starting dose of 50 mg BID was reduced to 40 mg BID in cycle 6 due to treatment-related grade 4 neutropenia, which lasted for 7 d.

Prolonged disease control in a patient with pancreatic neuroendocrine tumor
Discussion
This phase 1 study assessed the safety and PKs of the AAK inhibitor, alisertib, administered as an ECT formulation in patients with non-hematologic malignancies. The RP2D of the ECT formulation of single-agent alisertib in this patient population was determined to be 50 mg BID for 7 d followed by a 14-day rest period in 21-day cycles, similar to the RP2D determined in previous phase 1 studies of the PIC formulation of alisertib [21, 22].
Alisertib was generally tolerated in this patient population. Infrequent CNS AEs included hypersomnia, memory loss, and dizziness, and were presumed to be possible off-target effects related to the benzodiazepine structure of alisertib; these events were generally grades 1–2 in severity and were manageable and reversible. Neutropenia, febrile neutropenia, and leukopenia were the only drug-related AEs of grade 3 or higher reported in more than 2 patients. The most common non-hematologic AEs of alopecia and gastrointestinal toxicities most likely reflect the AAK inhibitory activity of alisertib in highly proliferative tissues. The safety profile observed in our study was largely consistent with previously reported clinical experience using the PIC formulation of alisertib [21, 22]. Neutropenia was the most frequent grade 3 or higher drug-related AE for each formulation across the three studies, and similar AEs were observed, including alopecia, hematological AEs, and gastrointestinal AEs. Similarly, in a phase 2 study that evaluated the PIC formulation of alisertib in patients with ovarian cancers [23], the most frequent drug-related AEs echoed the toxicity findings presented here for alisertib ECT.
The PK profile of alisertib ECT is also comparable to that observed in previous dose-escalation studies using the PIC formulation [21, 22]. Furthermore, in a relative bioavailability sub-study comparing the PIC and ECT formulations at 40 mg BID for 7 d [22, 24], systemic exposures were generally similar. The estimated relative bioavailability (geometric mean ratio of steady state alisertib AUC0-τ) of the ECT formulation in reference to the PIC is 90 % (90 % CI: 74.4–108.8 %; n = 14) [24]. Geometric mean steady-state trough concentrations at the RP2D of alisertib 50 mg BID administered as ECT exceeded 1 μM – the concentration associated with AAK inhibition and antitumor activity in preclinical xenograft models [27]. Our data support the conclusions from previous analyses and the transition to ECT in future clinical studies.
Previous phase 1 and 2 trials have showed that alisertib treatment was associated with durable disease control [21–23, 27]. In the present phase 1 study, no complete or partial responses by RECIST were observed. Ongoing phase 2 studies may yield additional information regarding response rates and progression-free survival in specific tumor types, as well as long-term safety information.
In conclusion, in adults with non-hematologic malignancies, the MTD and RP2D for alisertib ECT were determined to be 50 mg BID × 7 d, followed by a 2-week treatment-free period in 21-day cycles. This dose and schedule is evaluated in the phase 2 portion of the study (NCT01045421) in patients with NSCLC, SCLC, head and neck, gastroesophageal, and breast cancers. In parallel, additional single agent and combination studies of alisertib using the ECT formulation are ongoing in patients with solid tumors and hematologic malignancies.
References
Bolanos-Garcia VM (2005) Aurora kinases. Int J Biochem Cell Biol 37:1572–1577
Barr AR, Gergely F (2007) Aurora-A: the maker and breaker of spindle poles. J Cell Sci 120:2987–2996
Carvajal RD, Tse A, Schwartz GK (2006) Aurora kinases: new targets for cancer therapy. Clin Cancer Res 12:6869–6975
Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, Saya H (2003) Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 278:51786–51795
Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, Ecsedy J, Manfredi MG, Hyer ML (2010) MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res 8:373–384
Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, Santo L, Tai YT, Nahar S, Zheng M, Bandi M, Carrasco RD, Raje N, Munshi N, Richardson P, Anderson KC (2010) A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 115:5202–5213
Fu J, Bian M, Jiang Q, Zhang C (2007) Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 5:1–10
Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y (2008) Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res 14:4455–4462
Ogawa E, Takenaka K, Katakura H, Adachi M, Otake Y, Toda Y, Kotani H, Manabe T, Wada H, Tanaka F (2008) Perimembrane Aurora-A expression is a significant prognostic factor in correlation with proliferative activity in non-small-cell lung cancer (NSCLC). Ann Surg Oncol 15:547–554
Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL (2009) Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck 31:625–634
Zhang XH, Rao M, Loprieato JA, Hong JA, Zhao M, Chen GZ, Humphries AE, Nguyen DM, Trepel JB, Yu X, Schrump DS (2008) Aurora A, Aurora B and survivin are novel targets of transcriptional regulation by histone deacetylase inhibitors in non-small cell lung cancer. Cancer Biol Ther 7:1388–1397
Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, Yuan J, Ye J, Ozeck M, Shi ST, Zheng X, Rejto PA, Kan JL, Christensen JG, Pavlicek A (2012) An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Mol Cancer Ther 11:710–719
Hoque A, Carter J, Xia W, Hung MC, Sahin AA, Sen S, Lippman SM (2003) Loss of aurora A/STK15/BTAK overexpression correlates with transition of in situ to invasive ductal carcinoma of the breast. Cancer Epidemiol Biomarkers Prev 12:1518–1522
Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W (2008) Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer 112:1688–1698
Pan JY, Ajani JA, Gu J, Gong Y, Quin A, Hung M, Wu X, Izzo JG (2012) Association of Aurora-A (STK15) kinase polymorphisms with clinical outcome of esophageal cancer treated with preoperative chemoradiation. Cancer 118:4346–4356
Yang SB, Zhou XB, Zhu HX, Quan LP, Bai JF, He J, Gao YN, Cheng SJ, Xu NZ (2007) Amplification and overexpression of Aurora-A in esophageal squamous cell carcinoma. Oncol Rep 17:1083–1088
Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, Germanos MS, Bolen JB, Claiborne CF, Sells TB (2011) Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17:7614–7624
Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, Gorlick R, Kolb EA, Keir ST, Wu J, Wozniak AE, Yang Y, Manfredi M, Ecsedy J, Wang J, Neale G, Houghton PJ, Smith MA, Lock RB (2011) Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol 68:1291–1304
Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J, Smith MA, Houghton PJ (2010) Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer 55:26–34
Tomita M, Mori N (2010) Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci 101:1204–1211
Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, Rosello S, Andreu J, Jung J, Sanchis-Garcia JM, Piera A, Blasco I, Manos L, Perez-Fidalgo A, FIngert H, Baselga J, Tabernero J (2012) Phase 1 pharmacokinetic and pharmacodynamic study of MLN8237 (alisertib) – an investigational, oral, selective aurora a kinase inhibitor – in patients with advanced solid tumors. Clin Cancer Res 18:4764–4774
Dees EC, Cohen RB, Von MM, Stinchcombe TE, Liu H, Venkatakrishnan K, Manfredi M, Fingert H, Burris HA, Infante JR (2012) Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res 18:4775–4784
Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X, Danaee H (2012) Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol 127:63–69
Venkatakrishnan K, Infante J, Cohen R, Cohen RB, Burris HA, Zhou X, Liu H, FIngert H, Dees EC (2011) Phase 1b relative bioavailability (BA) study of enteric coated tablet (ECT) in reference to powder in capsule (PIC) formulation of the investigational drug MLN8237, an Aurora A kinase (AAK) inhibitor, in patients with advanced nonhematologic malignancies. Mol Cancer Ther 10:Abstract C122
National Cancer Institute. Common Terminology Criteria for Adverse Events v4.0. NCI, NIH, DHHS. [(2009) NIH publication # 09–7473. Available from URL: http://evs.nci.nih.gov/ftp1/CTCAE/About.html
Eisenhauer EA, Therasse P, Bogaerts J, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Friedberg JW, Mahadevan D, Cebula E, Persky D, Lossos I, Agarwal AB, Jung J, Burack R, Zhou X, Leonard EJ, Fingert H, Danaee H, Bernstein SH (2014) Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol 32:44–50
Acknowledgments
The authors would like to thank the patients who participated in these studies and their families, as well as staff at all investigational sites. Dr Sunil Sharma would like to acknowledge Teri Richards for administrative support.
The authors would also like to acknowledge Catherine Crookes and Nadia Korfali of FireKite for writing assistance in the development of this manuscript, which was funded by Millennium: The Takeda Oncology Company. The authors would like to express additional thanks to Adrienne Howard and Rosa Mostorino.
This work was performed in the U.T. M.D. Anderson Cancer Center Clinical and Translational Research Center (CTRC) and was supported by the Center for Clinical and Translational Sciences, which is funded by National Institutes of Health Clinical and Translational Science Award UL1 RR024148 and by the National Institutes of Health Cancer Center Support Grant (CCSG) award CA016672 to MD Anderson Cancer Center. The project described was performed at Huntsman Cancer Institute, which is supported by Award Number P30CA042014 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Conflict of interest
Research funding: SS, GF (Millennium: The Takeda Oncology Company)
Employment: XZ, HS, HF (Takeda Pharmaceuticals International Co.)
Equity ownership: SS (Beta Cat Pharmaceuticals, Salarius, ConverGene)
No conflicts of interest to disclose: RK, LG, DH, KM
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Falchook, G., Kurzrock, R., Gouw, L. et al. Investigational Aurora A kinase inhibitor alisertib (MLN8237) as an enteric-coated tablet formulation in non-hematologic malignancies: Phase 1 dose-escalation study. Invest New Drugs 32, 1181–1187 (2014). https://doi.org/10.1007/s10637-014-0121-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-014-0121-6