
Summary
A phase I trial of first-line vorinostat, an orally bio-available histone deacetylase inhibitor, in combination with capecitabine plus cisplatin (XP) was performed to assess recommend phase II trial dose in patients with advanced gastric cancer. Five dose levels of three-weekly vorinostat-XP were tested; vorinostat was dosed at 300–400 mg once daily on Days 1–14, capecitabine at 800–1,000 mg/m2 twice daily on Days 1–14, and cisplatin at 60–80 mg/m2 on Day 1. To assess the pharmacodynamics of vorinostat, histone H3 acetylation was assessed in peripheral blood mononuclear cells before the study treatment and at Day 8 of cycle 1. In total, 30 patients with unresectable or metastatic gastric adenocarcinoma were included. Dose-limiting toxicities were thrombocytopenia, fatigue, stomatitis, and anorexia. The following doses were recommended for phase II trial: 400 mg of vorinostat once daily, 1,000 mg/m2 of capecitabine twice daily, and 60 mg/m2 of cisplatin. The most common grade 3–4 toxicities were neutropenia (47 %), anorexia (20 %), thrombocytopenia (17 %), and fatigue (13 %). In overall, response rate was 56 % (95 % confidence interval [CI]: 32–81). With a median follow-up of 14.1 months, the median progression-free survival and overall survival were 7.1 months (95 % CI: 3.8–10.3) and 18.0 months (95 % CI: 4.8–31.1), respectively. The change in H3 acetylation after treatment with vorinostat correlated significantly with the vorinostat dose (300 vs. 400 mg/day) and the baseline level of H3 acetylation before treatment. Three-weekly vorinostat-XP regimen is feasible and recommended for further development in advanced gastric cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is the second leading cause of cancer-related deaths worldwide [1]. In unresectable or metastatic disease, palliative chemotherapy improves quality of life and survival outcomes compared to the best supportive care alone [2]. Combination chemotherapy regimens consisting of fluoropyrimidines and platinum are the standard therapeutic option in advanced gastric cancer [2–5]; however, the prognosis remains dismal and greater efficacy is needed.
Vorinostat (Zolinza®, Merck & Co., Inc.) is an orally bio-available histone deacetylase (HDAC) inhibitor that alters the level of acetylation of histone and non-histone proteins involved in the regulation of gene expression, angiogenesis, cell proliferation, and cell survival [6]. Previous studies showed that HDAC inhibitors induce growth arrest, terminal differentiation, apoptosis, or autophagic cell death in tumor cells by post-translational epigenetic modifications [6]. In addition, HDAC inhibitors have additive or synergistic effects when combined with other anticancer agents [7]. Vorinostat is currently approved for the treatment of refractory cutaneous T-cell lymphoma [8, 9] and has been investigated in several types of cancer, both as monotherapy and in combination with other agents, including 5-fluorouracil and platinum [10–13].
Previous studies suggested that the prognosis of patients with gastric cancer was associated with levels of HDAC expression and global histone modification [14, 15]. Furthermore, HDAC inhibition suppresses the growth of gastric cancer cells in vitro [16]. Based on these findings, we explored the role of vorinostat in unresectable or metastatic gastric cancer. We report the results of a phase I dose-finding and pharmacodynamics study of vorinostat combined with capecitabine plus cisplatin (XP), a current standard first-line regimen that has demonstrated non-inferiority to 5-fluorouracil and cisplatin in terms of efficacy and safety [3].
Materials and methods
Patient eligibility
Patients with histologically-documented unresectable or metastatic gastric adenocarcinoma were eligible if they met the following inclusion criteria: aged between 18 and 70 years; Eastern Cooperative Oncology Group (ECOG) performance status of 0–2; completion of adjuvant chemotherapy 6 months before the entry of study or no previous chemotherapy; adequate bone marrow, renal and liver function; no prior radiotherapy; and estimated life expectancy of >3 months. Patients were excluded if they had gastric outlet or intestinal obstruction, evidence of gastrointestinal bleeding, or received adjuvant treatment with capecitabine or platinum. Prior exposure to any HDAC inhibitor was not permitted for enrollment, but previous administration of valproic acid was allowed if a 30 day wash-off period was provided. The protocol was approved by the Institutional Review Board of Asan Medical Center, Seoul, Korea, and all patients provided written informed consent before enrollment. The study was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice (ClinicalTrial.gov Identifier: NCT01045538).
Study design and treatment plan
This study is a single-center, phase I dose-finding study with a safety expansion arm to define the maximum tolerated dose (MTD) and recommended phase II dose (RD) of vorinostat-XP regimen in patients with unresectable or metastatic advanced gastric cancer. The standard 3+3 dose escalation scheme was used, and a minimum of three patients were treated at each dose level. The dosing scheme for oral capecitabine (Days 1–14), intravenous cisplatin (Day 1) and oral vorinostat (Days 1–14) is presented in Table 1. The inclusion of patients in dose levels 2A and 2B proceeded simultaneously because we could not predict which level would be more toxic. Capecitabine was given twice daily. Cisplatin was administered in 150 ml of normal saline over 60 min with adequate pre- and post-hydration and premedication (steroids and antiemetics). Vorinostat was given once daily with food approximately 1 h before the administration of capecitabine. Prophylactic use of colony-stimulating factors was not permitted. Treatment continued until evidence of progression, unacceptable toxicity, or patient request for withdrawal. Patients received up to eight cycles of study treatment, after which, if they had not progressed, they received a combination of vorinostat and capecitabine without cisplatin at the same dose and schedule until disease progression or intolerable toxicity.
The MTD was determined based on the toxicity data from the first cycle. Dose-limiting toxicities (DLTs) were defined as follows: granulocytes <500/μl for more than 5 days; granulocytes <1,000/μl with fever; or grade 4 thrombocytopenia or any other non-hematologic grade 3/4 toxicity (excluding alopecia) that did not improve to at least grade 1 within 2 days of instituting the appropriate therapy, or that led to the interruption of capecitabine or vorinostat, resulting in the patient missing >25 % of the prescribed dose within a cycle, or to delays of more than 14 days in initiating the subsequent cycle because of persistent grade 2 or higher toxicity. Dose escalation continued until two or more of six patients experienced DLTs; that dose level was defined as MTD. Intra-patient dose escalation was not permitted. In patients who experienced DLT, re-challenge with chemotherapy was conducted with one level below the previous dose. The RD for phase II study was defined as the dose level below the MTD. RD was tested in an expansion cohort (n = 6) to confirm toxicity and safety.
Dose modifications
Before the start of every cycle of chemotherapy, patients were required to have absolute neutrophil counts of ≥1,500/μl and platelet counts of ≥100,000/μl; otherwise, the treatment was delayed by up to 3 weeks until recovery. In patients who experienced grade 4 neutropenia for >5 days, grade 4 thrombocytopenia, or grade ≥3 febrile neutropenia, doses of capecitabine and cisplatin were reduced by 25 % at the subsequent cycle. Capecitabine was interrupted for grade ≥2 non-hematologic toxicities with appropriate therapy, and missed doses were not replaced. If non-hematologic toxicities were resolved or decreased to grade 1, patients were re-challenged with capecitabine with a dose reduced up to 50 %, depending on the number and severity of the toxicities observed as specified in the protocol. The dose of cisplatin was modified if creatinine clearance was decreased or grade ≥3 nausea/vomiting and grade ≥2 neurotoxicity occurred. For hematologic toxicities, the dose of vorinostat was reduced (100 mg reduction) if grade 3 toxicity occurred; for grade 4 toxicity, vorinostat was interrupted until recovery to grade ≤2 and restarted at a reduced dose. For non-hematologic toxicities, vorinostat was delayed and restarted at a reduced dose when the toxicity improved to grade ≤1 for grade 3 toxicity, and discontinued for grade 4 toxicity. If more than two dose reductions were required, vorinostat was stopped.
Assessment of response and toxicity
Prior to entering the study, patients underwent baseline assessments, including history, physical examination, complete blood count (CBC) with differential counts, serum chemistry, electrolytes, determination of coagulation parameters, urinalysis, electrocardiography, chest X-ray, and computed tomography (CT) scanning of the abdomen and pelvis. Other investigations, such as bone scan and chest CT scan, were performed if there was metastatic disease. Physical examinations, chest X-rays, CBC, chemistry, and electrolytes were repeated prior to each chemotherapy cycle. CBC was performed weekly until completion of the 2nd cycle. Tumor response was evaluated every 2 cycles according to RECIST criteria version 1.0 using the same imaging technique as that used at baseline [17]. Patients with documented progressive disease were monitored every 3 months until death. Adverse events were evaluated before each treatment cycle according to the National Cancer Institute Common Terminology Criteria, version 3.0. All patients were reviewed approximately 1 month after the last cycle of treatment to document any delayed adverse event.
Progression-free survival (PFS) and overall survival (OS) were estimated using the Kaplan–Meier method. PFS was measured from the start of treatment to document tumor progression or death from any cause, whichever occurred first. OS was calculated from the start of treatment to the date of death from any cause.
Pharmacodynamics
Peripheral blood (10 ml) was obtained from patients before the study treatment and 2 h after ingestion of vorinostat capsules at post-treatment Day 8 of cycle 1. The peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation using Lymphoprep (AXIS-SHIELD PoC, Oslo, Norway) according to the manufacturer’s instructions. Isolated PBMCs were lysed in M-PER buffer (Pierce, Rockford, IL, USA). Protein content was quantified using the Bradford assay (Bio-Rad, Hercules, CA, USA). Cell extracts separated by SDS-PAGE were transferred to nitrocellulose membrane (Millipore, Billerica, MA, USA) for Western blot analysis. The blots were incubated overnight at 4 °C with primary antibodies against histone acetyl-H3 (Cell Signaling, Danvers, MA, USA), HDAC2 (Cell Signaling) and β-actin (Sigma, St. Louis, MO, USA). The blots were stripped and re-probed for histone H3 (Cell Signaling) and images were quantified using the Multi-Gauge v2.3 software (Fujifilm, Tokyo, Japan). Histone H3 acetylation was measured by quantifying acetyl-H3 band pixel intensity and normalising it to the respective H3 band in a representative immunoblot. The change between pre- and post-vorinostat acetyl-H3 was calculated in each patient and described as percent change or fold increase.
Results
Patient characteristics
Between August, 2010, and June, 2011, a total of 30 patients (24 for a dose-finding study and 6 for an expansion cohort) were enrolled. Patient baseline characteristics are presented in Table 2. The median age was 50 years (range: 25–67) and half of the patients were male. Most patients (83 %) initially presented with metastatic disease. Metastasis in two or more organs was observed in 63 % of the patients. Among five patients who had recurrent disease, adjuvant chemotherapy had been given in four patients (13 %); doxifluridine and S-1 each for two patients. A total of 249 chemotherapy cycles (median: 8; range: 1–25) were administered. Twelve patients (40 %) completed the preplanned eight cycles of treatment, and 10 of them received the median eight cycles (range: 1–17) of further chemotherapy with capecitabine and vorinostat. All patients completed at least one cycle of chemotherapy and were available for safety assessments.
DLTs
The DLTs of the vorinostat-XP regimen were thrombocytopenia, fatigue, anorexia and stomatitis (Table 1). DLT was noted in one of six patients in level 1 (grade 4 thrombocytopenia), zero of three patients in level 2A, one of six patients in level 2B (grade 3 fatigue), one of six patients in level 3 (grade 3 stomatitis), and two of three patients in level 4 (grade 4 thrombocytopenia, and discontinuation of capecitabine or vorinostat for more than 25 % of the prescribed dosage due to grade 3 anorexia and fatigue). According to the protocol, dose level 4 was the MTD, so the RD was determined to be dose level 3 (400 mg of vorinostat once daily on Days 1–14, 1,000 mg/m2 of capecitabine twice daily on Days 1–14, 60 mg/m2 of cisplatin on Day 1, every 3 weeks). In the expansion cohort at level 3, DLTs did not occur.
Adverse events
The toxicity profile is summarized in Table 3. The most common grade 3–4 adverse events were neutropenia (47 %), anorexia (20 %), thrombocytopenia (17 %), and fatigue (13 %). Febrile neutropenia was not observed, and there was no treatment-related death. In patients who received RD (level 3), neutropenia (42 %) and anorexia (25 %) were the most frequent grade 3–4 toxicities, and the mean relative dose intensity (RDI, the total delivered dose as a percentage of the targeted dose per unit time) was maintained at least at 70 % for vorinostat, 66 % for capecitabine, and 71 % for cisplatin within the sixth cycle of study treatment (Fig. 1.)

Relative dose intensity of capecitabine, cisplatin, and vorinostat at the recommended dose (dose level 3)
Efficacy
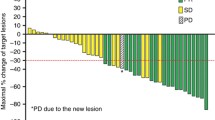
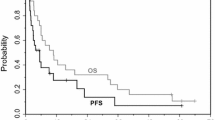
In overall, 18 patients had measurable disease, and two patients were not evaluable for response due to follow-up loss before the first response assessment. Partial response was observed in nine patients, providing an overall response rate of 56 % (95 % confidence intervals [CI], 32–81). Five patients (31 %) had stable disease and two patients (13 %) had progressive disease as best response. No patient achieved complete response. In level 3 cohort, who received RD, two (33 %) of six patients with measurable lesions showed responses, while three patients (50 %) and one patient (17 %) had stable disease and progressive disease, respectively. In six (level 1), five (level 2B), and one (level 4) patients with measurable lesions, tumor response was assessable in five, four, and one patients, respectively. Partial response was reported in four (67 %) of level 1 cohort and three (60 %) of level 2B cohort, and each patient in level 1 and level 4 cohorts had stable disease. In level 2A cohort, there was no patient who had measurable disease; therefore, response rate was not assessable. At the median 14.1 months (range: 5.0–22.7) of follow-up for surviving patients, the median PFS and OS were 7.1 months (95 % CI: 3.8–10.3), and 18.0 months (95 % CI: 4.8–31.1), respectively (Fig. 2).

Progression-free survival and overall survival. Median progression-free survival and overall survival were 7.1 months (95 % CI: 3.8–10.3) and 18.0 months (95 % CI: 4.8–31.1), respectively
Pharmacodynamic analysis
Pre- and post-vorinostat PBMCs were obtained in all patients except one. The median change in histone H3 acetylation between pre- and post-vorinostat samples was 9.1 % (95 % CI: −10.3–224.1; Fig. 3a). Although this was not significant overall (p = 0.09), H3 acetylation was significantly increased in patients who received 400 mg/day vorinostat (median 24.2 %, p = 0.02), but not in those who received 300 mg/day (4.0 %, p = 0.33). Moreover, the baseline level of H3 acetylation correlated significantly with the extent of the change observed after vorinostat treatment (p < 0.001; Fig. 3b). However, HDAC2 expression in PBMCs at baseline was not associated with the extent of the change in H3 acetylation (p = 0.38). Multivariate analysis was performed with the inclusion of the baseline HDAC2 level, baseline H3 acetylation level, and vorinostat dose (300 mg/day vs. 400 mg/day). Results show that the baseline H3 acetylation level (standardized coefficient, beta = −0.71, p < 0.001) and the dose of vorinostat (beta = 0.33, p = 0.03) correlated significantly with the change in H3 acetylation.

Percent change in histone H3 acetylation (a) and relationship between baseline H3 acetylation and change in H3 acetylation after treatment with vorinostat (b). In panel a, three patients had changes in acetyl-H3 greater than 200 % (1679.4 %, 342.6 %, and 419.6 %). In panel b, the fold increase in acetyl-H3 was log-transformed
Discussion
This phase I trial evaluated the three-weekly vorinostat-XP regimen as first-line chemotherapy in unresectable or metastatic gastric cancer. The results defined the RD as 400 mg of vorinostat once daily on Days 1–14, 60 mg/m2 of cisplatin on Day 1, and 1,000 mg/m2 of capecitabine twice daily on Days 1–14 for further clinical investigation. The DLTs of this combination regimen included grade 4 thrombocytopenia, grade 3 fatigue, grade 3 anorexia, and grade 3 stomatitis.
The adverse events are consistent with the toxicities previously reported in clinical trials of vorinostat alone and of the XP regimen alone [3, 9]. Adverse events were generally mild to moderate in severity. The most common grade 3–4 adverse events were neutropenia (47 %), anorexia (20 %), thrombocytopenia (17 %), and fatigue (13 %). In previous phase II trials of vorinostat monotherapy using a 400 mg once daily dosing schedule, thrombocytopenia (5–8 %) and fatigue (5 %) were the most common grade 3–4 adverse events [9, 18]. Neutropenia (16 %) and vomiting (7 %) were the most frequent grade 3–4 toxicities in a previous phase III trial of three-weekly XP regimen consisting of 1,000 mg/m2 of capecitabine twice daily on Days 1–14 and 80 mg/m2 of cisplatin on Day 1 [3]. The frequency of severe hematologic toxicities, such as neutropenia and thrombocytopenia, was much higher than that observed in a previous phase III trial of XP, likely because of the added toxicity of vorinostat; however, this increase in frequency might be an overestimation considering that CBC was monitored every week until completion of the 2nd cycle in this study and every 3 weeks in the previous phase III trial of XP [3]. Considering that the RDIs of each drug were well maintained through the course of treatment, most of the toxicities of the vorinostat-XP regimen were tolerable and manageable.
The objective response was 56 %, and the median PFS and OS were 7.1 months and 18.0 months, respectively. These efficacy results are comparable to those of previous trials for first-line chemotherapy in advanced gastric cancer; however, considering that patients received different doses of chemotherapy and only half of the patients were available for response assessment, it is premature to compare the efficacy of the vorinostat-XP regimen to that of other regimens. Efficacy should be validated in future trials.
In the pharmacodynamics analysis, the change in H3 acetylation after treatment with vorinostat was comparable to that observed in previous studies [19, 20]. Although a previous study had failed to show any correlation between vorinostat dose and histone H3 acetylation in PBMCs [19], our results demonstrated that the increase in H3 acetylation was more prominent in patients who received 400 mg/day vorinostat than in those who received 300 mg/day. This indicates that 400 mg/day vorinostat, the RD in the vorinostat-XP regimen, might be more appropriate than 300 mg/day from a pharmacodynamic viewpoint. Furthermore, results showed that a lower baseline H3 acetylation level was associated with greater increase of H3 acetylation level after vorinostat. The effects of baseline H3 acetylation and vorinostat dose remained significant by multivariate analysis. Considering that there is no established bio-marker for prediction of efficacy and toxicity in patients treated with vorinostat, baseline H3 acetylation in PBMCs should be investigated in future trials as a potential bio-marker.
In the era of targeted therapy, various biologic agents have been investigated for the treatment of advanced gastric cancer in combination with conventional chemotherapy regimens in previous studies [21–23]; however, so far, only trastuzumab combined with XP or 5-fluorouracil plus cisplatin improved survival significantly in HER2-overexpressing gastric cancer [21]. Therefore, the development of new agents to enhance the efficacy of current standard regimens in advanced gastric cancer is urgently needed. Our results provide a compelling rationale for further clinical investigation of the vorinostat-XP regimen in gastric cancer.
In conclusion, vorinostat-XP is a feasible first-line therapy regimen in advanced gastric cancer. A phase II trial based on the results of the present study is now ongoing. Further clinical trials are warranted to evaluate bio-markers as well as the safety and efficacy of this regimen.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61(2):69–90
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE (2006) Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol 24(18):2903–2909
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L (2009) Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 20(4):666–673
Koizumi W, Narahara H, Hara T, Takagane A, Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama W (2008) S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol 9(3):215–221
Kang YK, Yoon DH, Ryoo BY, Ryu MH (2010) Recent advances in chemotherapy for advanced gastric cancer. Asia Pac J Oncol Hematol 2(1):67–74
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5(9):769–784
Marks P, Richon V, Miller T, Kelly WK (2004) Histone deacetylase inhibitors. Adv Cancer Res 91:137–168
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12(10):1247–1252
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109(1):31–39
Fakih MG, Fetterly G, Egorin MJ, Muindi JR, Espinoza-Delgado I, Zwiebel JA, Litwin A, Holleran JL, Wang K, Diasio RB (2010) A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin Cancer Res 16(14):3786–3794
Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, Espinoza-Delgado I, Litwin A, Rustum YM, Ross ME, Holleran JL (2009) A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin Cancer Res 15(9):3189–3195
Ramalingam SS, Parise RA, Ramananthan RK, Lagattuta TF, Musguire LA, Stoller RG, Potter DM, Argiris AE, Zwiebel JA, Egorin MJ (2007) Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res 13(12):3605–3610
Vansteenkiste J, Van Cutsem E, Dumez H, Chen C, Ricker JL, Randolph SS, Schöffski P (2008) Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Investig New Drugs 26(5):483–488
Park YS, Jin MY, Kim YJ, Yook JH, Kim BS, Jang SJ (2008) The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann Surg Oncol 15(7):1968–1976
Weichert W, Röske A, Gekeler V, Beckers T, Ebert MPA, Pross M, Dietel M, Denkert C, Röcken C (2008) Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol 9(2):139
Park JH, Jung Y, Kim TY, Kim SG, Jong HS, Lee JW, Kim DK, Lee JS, Kim NK, Kim TY (2004) Class I histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin Cancer Res 10(15):5271–5281
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25(21):3109–3115
Munster P, Marchion D, Thomas S, Egorin M, Minton S, Springett G, Lee J, Simon G, Chiappori A, Sullivan D (2009) Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer 101(7):1044–1050
Fujiwara Y, Yamamoto N, Yamada Y, Yamada K, Otsuki T, Kanazu S, Iwasa T, Hardwick JS, Tamura T (2009) Phase I and pharmacokinetic study of vorinostat (suberoylanilide hydroxamic acid) in Japanese patients with solid tumors. Cancer Sci 100(9):1728–1734
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376(9742):687–697
Ohtsu A, Shah MA, Van Cutsem E, Rha SY, Sawaki A, Park SR, Lim HY, Yamada Y, Wu J, Langer B (2011) Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: a randomized, double-blind, placebo-controlled phase III study. J Clin Oncol 29(30):3968–3976
Waddell TS, Chau I, Barbachano Y, de Castro DG, Wotherspoon A, Saffery C, Middleton GW, Wadsley J, Ferry DR, Mansoor W (2012) A randomized, multicenter trial of epirubicin, oxaliplatin, and capecitabine (EOC) plus panitumumab in advanced esophagogastric cancer (REAL3). J Clin Oncol 30(suppl; abstr LBA4000)
Acknowledgment
Merck & Co., Inc. generously provided vorinostat and funded part of the study.
Conflict of interest
Yoon-Koo Kang received a research grant from Merck & Co., Inc., and honoraria for a lecture and a research grant from Roche. Besides, there is no conflict of interest to be reported from other authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yoo, C., Ryu, MH., Na, YS. et al. Phase I and pharmacodynamic study of vorinostat combined with capecitabine and cisplatin as first-line chemotherapy in advanced gastric cancer. Invest New Drugs 32, 271–278 (2014). https://doi.org/10.1007/s10637-013-9983-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9983-2