
Summary
CP-4126 is a gemcitabine (2′,2′-difluorodeoxycytidine; dFdC) 5′ elaidic acid ester. The purpose of this dose-escalating study was to assess safety, pharmacokinetics (PK) and preliminary antitumor activity of the oral formulation and to determine the recommended dose (RD) for phase II studies. The study had a two-step design: a non-randomized dose-escalating step I with oral CP-4126 alone, followed by a randomized, cross-over step II that compared oral CP-4126 with dFdC i.v.. CP-4126 was given on days 1,8,15 in a 4-week schedule with increasing doses until the RD was established. 26 patients with different solid tumours were enrolled in step I at seven dose levels (100–3,000 mg/day). The most frequent drug-related AEs were fatigue and dysgeusia, the majority being grade 1–2. One patient experienced a dose limiting toxicity after one dose of CP-4126 at 1,300 mg/day (ASAT grade 3). PK of CP-4126 could not be determined. The metabolites dFdC and dFdU obeyed dose-dependent pharmacokinetics. Exposures to dFdC were about ten-fold lower compared to exposures after comparable doses of dFdC i.v.. Nine patients reached stable disease as best response, whereby in one patient with vaginal carcinoma a 25 % reduction of tumor volume was reached. This study demonstrates that CP-4126 can be safely administered orally to patients up to 3,000 mg/day in a d1,8,15 q4w schedule with a tolerable safety profile. CP-4126 acts as a prodrug for dFdC when given orally, but because of the poor absorption and the rapid pre-systemic metabolism the study was terminated early and no RD could be determined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CP-4126 is a gemcitabine (2′,2′-difluorodeoxycytidine; dFdC) 5′ elaidic acid ester. dFdC is a nucleoside analogue used in the treatment of patients with various solid tumors, including non-small cell lung cancer and pancreatic carcinoma [1]. dFdC is currently formulated as an i.v. solution and usually administered as 30-minute infusion at a dose of 1,000 to 1,250 mg/m2 on days 1 and 8 of a 21-day or days 1, 8, and 15 of a 28-day cycle. dFdC is a hydrophilic compound, which needs to be actively transported into cells by human equilibrative and concentrative nucleoside transporters (hENT and hCNT) to exert its anticancer activity with hENT1 being the primary transporter [2]. A mechanism of resistance to dFdC is a decreasing transport activity of nucleosides across the cell membrane [2–4].
When dFdC has been transported into the cell, it is phosphorylated by deoxycytidine kinase (dCK) to its active diphosphate and triphosphate forms (respectively dFdCDP and dFdCTP) [3, 5]. dFdCDP slows the synthesis and repair of DNA by inhibition of ribonucleotide reductase [6], which also subsequently leads to an increase in dCK activity. The other active form, dFdCTP, competes with deoxycytidinetriphosphate for incorporation into DNA, thereby inhibiting DNA polymerase and preventing the activity of DNA repair enzymes [7] (see Fig. 1). At high concentrations, it also directly inhibits deoxycytidine monophosphate deaminase, resulting in decrease in gemcitabine catabolism [8]. The main metabolite of dFdC, 2′,2-difluorodeoxyuridine (dFdU), is formed by deoxycytidine deaminase, which is present at high levels in plasma, red blood cells and liver [9, 10].

Cellular uptake, activation and deactivation of CP-4126
Besides the general advantages of an intravenous-to-oral switch in anticancer chemotherapy [11], oral gemcitabine has a potential to be applied on a low-dose metronomic treatment regime [12, 13]. Several preclinical studies have shown that gemcitabine has anti-tumor and anti-angiogenic properties when used chronically and continuously at low-daily dose [14, 15].
There are two previously published clinical studies with an oral dosage form of gemcitabine, one of which with oral administration of gemcitabine itself [16]. This study was terminated because of high presystemic conversion of dFdC to dFdU and accompanying accumulation of dFdU, which most likely contributed to severe liver toxicity. The second dosage form tested orally was the prodrug LY2334737, which was designed to overcome the pre-systemic deamination of gemcitabine during first pass metabolism [17]. LY2334737 has valproic acid bound to the metabolically unstable amine group and is therefore considered not to be a substrate for deaminase [18].
CP-4126 was originally developed as an intravenous formulation. It was synthesized in order to improve the clinical activity of gemcitabine [19]. An elaidic fatty acid (trans-9-octadecenoic fatty acid) was esterified at the 5′position of dFdC, which gives CP-4126 two advantages over dFdC. The first advantage is that unlike dFdC, CP-4126 can traverse cell membranes by passive diffusion, followed by intracellular conversion to gemcitabine by esterases. This means that uptake of CP-4126 in the cell is independent of nucleoside transporters and thereby relatively independent of multidrug resistance mechanisms mediated by down regulation of hENT and hCNT expression [20]. Since hENTcan operate as export as well as uptake transporters, the clinical activity could be improved by cellular accumulation and prolonged retention of the active metabolites [21]. Secondly, like LY2334737, CP-4126 appears not to be a substrate for deaminase [20], although the fatty acid is bound to the 5′position of dFdC and not to the metabolically unstable amine group. In a preclinical study in dogs, CP-4126 could be given orally, where it acts as a prodrug for gemcitabine with a high apparent systemic availability of dFdC [20]. Because of these potential advantages, the aim of this study was to explore whether oral CP-4126 is a prodrug of dFdC and whether it is possible to achieve pharmacologically significant systemic exposure to dFdC after oral administration of CP-4126.
In this dose escalation phase I study, patients with advanced solid tumors were treated with oral CP-4126. The primary objective was to determine the recommended dose for phase II studies (RD) of oral CP-4126. The secondary objectives were to determine the safety, pharmacokinetics (PK) of CP-4126, dFdC and dFdU, and preliminary antitumor activity of the oral formulation.
Patients and methods
Eligibility
Patients with histologically or cytologically confirmed locally advanced or metastatic solid tumors, for whom there remained no known effective treatment, were eligible. Patients with symptomatic brain metastases were excluded. Patients needed to be 18 years of age or older and have a performance status 0–2 according to the Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) Scale. The life expectancy needed to be be longer than 3 months and the bone marrow, haematological and biological functions had to be adequate (neutrophil count of ≥1.5 * 109/L, platelets of ≥100 * 109/L, and hemoglobin of ≥10 g/dL; alanine transaminase (ALT) and aspartate transaminase (AST) ≤2.5 times institutional upper limit of normal (ULN) (if liver metastases, AST/ALT ≤5 times ULN and alkaline phosphatase ≤2.5 times ULN), bilirubin of ≤1.5 times of the ULN (liver metastases: ≤ 2 times of the ULN); serum creatinine ≤1.5 of the upper limit of the ULN). Main exclusion criteria were current peripheral neuropathy of grade 1 or higher, mucositis of the upper digestive tract, or if the patient had received previous anticancer therapy (chemotherapy, hormonal therapy or immunotherapy) within 30 days prior to the first dose of oral CP-4126 (6 weeks for mitomycin C and BCNU (= carmustine) and CCNU (=lomustine)). Also radiotherapy to more than 30 % of bone marrow or single dose radiation up to 8 Gy, less than 1 week prior to the study treatment or to the upper GI tract was an exclusion criterion. The study protocol was approved by the local Medical Ethics Committee and all patients had to give written informed consent.
Drug formulation
For the purpose of oral administration, CP-4126 was solubilised in a lipid-based formulation and encapsulated in non-gelatine hard-shell capsules for oral application. The capsules were provided by Clavis Pharma in two strengths: 100 mg and 250 mg.
Study design and treatment schedule
This phase I study was a first-in-human, non-randomized, multicentric, open-label, dose escalation study of oral CP-4126 as monotherapy. CP-4126 capsules were administered once a day on days 1,8,15 in a 4-week schedule (d1,8,15 q4w). CP-4126 capsules were taken in fasting conditions (i.e., in the morning, at least 2 h after and at least 1 h before food intake) since the effect of food on the pharmacokinetics of the drug was unknown. The starting dose was 100 mg. Three to six patients were enrolled at each dose-level (DL), according to predefined dose escalation decision rules. The doses were increased based on safety evaluations per dose-level. The dose limiting toxicities (DLTs) were defined as any of the following events occurring during the first treatment cycle and related to study treatment: non-hematologic AE of CTCAE grade ≥3 (excluding alopecia), nausea and vomiting (only after adequate systemic antiemetic medication), neutropenia CTCAE grade 4 (i.e., neutrophils <0.5 × 109/L, lasting ≥5 days), febrile neutropenia, (i.e., neutrophils <1.0 × 109/L with fever at least 38.5 °C), thrombocytopenia CTCAE grade 4 and treatment delay >1 week/cycle due to treatment related toxicity. The RD was defined as the highest DL where ≤1 of 6 patients experience a DLT.
Safety evaluation and treatment adjustments
Pre-treatment evaluation included a complete medical history, physical examination, ECG, vital signs, performance status, assessment of adverse events using CTCAE v3.0, the use of concomitant medications, pregnancy test, laboratory assessment of hematology, serum chemistry and urinalysis and a tumor assessment. Before each administration a physical examination, assessment of adverse events and notation of concomitant medication were repeated and hematology and serum chemistry were checked.
The next treatment cycle was allowed to be delayed for a maximum of 1 week (maximum duration of 5 weeks per cycle), and at least five out of the six dose administrations had to be given during the previous two cycles. In cases where subsequent treatment cycles were delayed for more than 1 week or if the patient had received less than five dose administrations within the previous two cycles the patient was withdrawn from the study. If the patient was obtaining clinical benefit and was able to continue treatment at a dose-level decided upon by the investigator, in agreement with the sponsor and the Project Medical Advisor (PMA), the patient was allowed to continue if in his/her best interest.
If a patient had experienced a DLT, further treatment was allowed at a lower dose if the patient fulfilled the inclusion/exclusion criteria, the investigator expected clinical benefit from further treatment, and the investigator, the PMA and the sponsor were in agreement.
Patients treated according to an adjusted treatment and/or modified dose were evaluated for safety and efficacy at this DL, but any DLTs that developed at the new DL were not added to the total number of DLTs at that dose-level or to the study.
Pharmacokinetics
The pharmacokinetics of CP-4126, dFdC and dFdU were monitored on Day 1 of the first cycle of the study. The sampling times were pre-treatment, 0.5, 1, 1.5, 2, 3, 4, 8 and 24 h. Blood samples were collected for study drug analysis in tubes containing the enzyme inhibitors potassium fluoride and tetrahydrouridine in addition to lithium heparin as anticoagulant. Plasma samples were measured by York Bioanalytical Solutions using a validated high-performance liquid chromatography tandem mass spectrometry (HPLC/MS/MS) method to determine the plasma concentrations of CP-4126, dFdC and dFdU. For the determination of CO-1.01, the analog CP-4055 was used as internal standard and ARA-U was used for the determination of dFdC and dFdU. Sample pre-treatment was done by protein precipitation with acetonitrile. A volume of 100 μL human plasma was processed. After mixing, samples were centrifuged and a part of the clear supernatant was diluted and submitted for the CP-4126 LC-MS/MS analysis. The other part was dried under a gentle stream of nitrogen. Residues were reconstituted with ammonium acetate and submitted for the dFdC/dFdU LC-MS/MS analysis. Detection was performed in positive ion mode on a tandem MS with a Turbo Ion Spray Interface (MDS Sciex API 3000). The transitions for CP-4126 were selected from m/z 528 to 264 and for the internal standard from m/z 508 to 112. The transitions for dFdC and dFdU were selected from m/z 264 to 112 and 265 to 113, respectively and for the internal standard from m/z 245 to 115. PK parameters, including Cmax, Tmax, AUC, and T1/2 were calculated using WinNonlin Pro version 5 (Pharsight Corporation, USA).
Anti-tumor activity
Tumor measurements were recorded using RECISTv1.1 [22]. Responses based on other markers, for example prostate-specific antigen (PSA), were also recorded.
Results
Patient characteristics and disposition
A total of 26 patients were enrolled in the dose-escalation study divided over seven dose-levels (100, 200, 400, 800, 1,300, 2,000 and 3,000 mg/day). The median age was 62 years (range, 42–72 years) and 88 % had an ECOG performance status ≤1. Almost 70 % were females and all patients were Caucasians (Table 1). All enrolled patients received at least one dose of CP-4126, of which the majority (88 %) completed at least one cycle. The median number of cycles entered on treatment for all patients in the seven dose-levels was 2 (range 1–4), 2, 2 (range 1–2), 2 (range 2–5), 2.5 (range 1–8), 4 (range 2–4), 1.5 (range 1–6) respectively. In total 21 patients (81 %) discontinued study treatment permanently due to progressive disease, the other patients discontinued treatment as a result of one or more adverse events (all ≥grade 3, one event at the 1,300 mg level was fatal). None of the patients discontinued due to toxicity related to CP-4126. Reductions were equally distributed among all dose-levels and there were no clear differences in dose delays and interruptions between the dose-levels.
The RD was not reached, as the study was stopped because of the poor absorption and subsequently low blood levels of the molecule.
Safety and tolerability
All patients treated (N = 26) were evaluated for treatment-related adverse events. Table 2 lists all adverse events which are possibly, probably or definitely related to the study drug with an incidence rate of at least 5 %. Oral CP-4126 at the dose levels studied was overall well tolerated. The most common drug-related adverse event was fatigue (38 %), the majority being grade 1–2. Also dysgeusia (19 %) and gastrointestinal disorders (mainly nausea and vomiting, 23 %) were often related to the study drug. There were no adverse events of neutropenia or thrombocytopenia reported during this study. One patient experienced a DLT after one dose of CP-4126 at 1,300 mg/day. The aspartate transaminase (AST) in this patient reached more than five times the upper limit of normal (CTCAE grade 3). Four patients experienced fatal adverse events of which two of the deaths occurred within 30 days after the last dose of CP-4126. All fatal adverse events were considered unrelated to the study drug. In three cases this was due to disease progression and in one case the patient developed a fatal event of dyspnoea approximately 2 months after starting treatment which also was considered unrelated to study drug. The death occurred approximately 35 days later.
Pharmacokinetic results
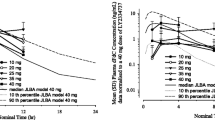
Pharmacokinetic blood samples were drawn from all patients on the first day of the study. PK profiles composed for dFdC and dFdU are shown in Figs. 2 and 3. Plasma concentrations of CP-4126 were only quantifiable in a few patients at the highest dose-levels around 2 h (range 1–4 h) post dose. There was no consistent relationship between the Cmax and the dose.

Mean plasma concentration-time curve of dFdC in patients treated with CP-4126 by oral capsule administration on day 1 of treatment

Mean plasma concentration-time curve of dFdU in patients treated with CP-4126 by oral capsule administration on day 1 of treatment
The observed Cmax and Tmax and calculated AUCt values for dFdC and dFdU per dose-level are presented in Table 3. The increase in systemic exposure of dFdC was proportional to the dose increase of CP-4126 (200 to 3000 mg). Also the systemic exposure of dFdU increased with increasing dose. However, these increases were less than the proportionate dose increase. For the majority of patients Cmax of dFdC and dFdU were reached at 1.4 h (range 0.5–4.1 h) and at 3.4 h (range 1–10.5 h) after dosing, respectively, independent of the dose. The mean terminal half-life of dFdC and dFdU could only be determined in the highest dose-levels and were respectively ~0.5 h and ~9.0 h.
Anti-tumor activity
Anti-tumor activity was a secondary endpoint for this study. No complete or partial responses were achieved. Nine patients reached stable disease as best response (35 %), whereby in one patient a 25 % reduction of tumor volume from baseline was reached (vaginal carcinoma). This patient had progression-free survival of more than 7 months.
Discussion
This report describes the first study of an oral formulation of CP-4126, an elaidic acid ester of gemcitabine. In this study the tolerability, safety and pharmacokinetics of CP-4126 were explored.
Seven dose-levels were evaluated in this study, ranging from 100 mg to 3000 mg. The treatment was overall well tolerated. Most reported adverse events were fatigue (38 %) and dysgeusia (19 %). The most commonly reported adverse drug reactions associated with i.v. treatment of gemcitabine are nausea with or without vomiting, elevated liver transaminases (AST/ALT) and alkaline phosphatase, all reported in approximately 60 % of patients [23]. This profile is not seen in this population at the dose levels studied, probably because of the lower exposures of dFdC compared to the exposures after i.v. treatment of gemcitabine.
Plasma concentrations of CP-4126 were only quantifiable in 16 of the 260 samples from 26 patients, without a consistent relationship between the dose of CP-4126 and the maximum concentrations or the exposures to CP-4126. The low amount of CP-4126 detected in plasma corresponds with PK data in dogs and indicates a rapid pre-systemic metabolism of this parent drug after oral administration [20]. This is probably due to the amount of carboxylesterases in liver and intestines, because these esterases are most likely responsible for the conversion of CP-4126 outside as well as inside the cell [24]. dFdC and dFdU concentrations were detectable in all patients. For both metabolites, the Cmax and exposure increased generally less than dose-proportionally with the dose, suggesting a capacity-limited absorption of CP-4126.
The exposures to dFdC (and dFdU) are significantly lower compared to the exposures after comparable doses of gemcitabine applied intravenously (on molar basis) [25, 26]. Although the comparison is made with patients from other studies, the exposure of dFdC seems to be about ten-fold lower after oral administration (apparent systemic availability of dFdC is 5–10 %). For dFdU the AUC is relatively higher, but still only 20–78 % in comparison with gemcitabine i.v.. These lower exposures after oral administration indicate low absorption of CP-4126 in humans. In dogs, however, oral CP-4126 acts as a prodrug for gemcitabine and the exposures of dFdC and dFdU were in the same range as the exposures after i.v. administration of CP-4126 (the apparent systemic availability is in the range of 92–169 %, depending on the dose) [20]. In dogs, the half-life of dFdC after oral administration of CP-4126 was longer than the half-life after i.v. administration of dFdC (unpublished data). In humans, the terminal half-life of dFdC after oral administration of CP-4126 is equal to the half-life after i.v. administration dFdC [25] and significantly shorter compared to the half-life after i.v. administration of CP-4126 (0.5 h versus 9 h [27]). These PK results indicate that a substantial amount of parent drug which is absorbed in the intestines is directly metabolized to dFdC and in consequence to dFdU. Due to the high levels of deoxycytidine deaminase in the liver, the relatively high concentrations of dFdU after the rapid hydrolysis of the elaidic acid of CP-4126 are expected. These results show a large pre-systemic metabolism of both CP-4126 and dFdC after oral administration.
Due to the low exposure of CP-4126 and gemcitabine, once weekly administration of CP-4126 is not feasible as a possible alternative for gemcitabine i.v.. A more chronic administration of CP-4126 could be an option as seen in preclinical studies [20]. Comparable treatment regimens are investigated with alternative oral dosage forms of gemcitabine in two other studies [16, 18]. In one study, oral gemcitabine was dosed every other day and the highest dose administered was 20 mg (equivalent to 40 mg CP-4126), before the study was terminated because of severe liver toxicity by dFdU accumulation. In another study, the prodrug LY2334737 was given once daily and the highest dose was 50 mg (equivalent to 70 mg CP-4126). A direct comparison with oral CP-4126 is difficult because of the difference in doses and treatment regimens, but the difference in the exposure ratio dFdU/dFdC between LY2334737 (range 216–404) and CP-4126 (range 261–1268) confirms the rapid pre-systemic metabolism of CP-4126 and dFdC (range after i.v. administration of dFdC is 31–201 [25]). Due to the long terminal half-life, dFdU accumulates during the first weeks after the metronomic treatment regimens. Although a clear causal relationship is lacking, the hepatotoxicity after chronic oral administration of both gemcitabine as LY2334737 is linked to the accumulation of dFdU and consequently the accumulation of phosphorylated dFdU (via deamination of gemcitabine monophosphate to dFdUMP and via phosphorylation of dFdU) in the liver, which can be incorporated into DNA and RNA [28]. The expected accumulation of dFdU by pre-systemic metabolism after more chronic administration of oral CP-4126 would make metronomic treatment regimens unsuitable.
In conclusion, this study demonstrates that CP-4126 can be safely administered orally to patients up to 3000 mg/day in a d1,8,15 q4w schedule. The best response was stable disease, which was seen in nine patients (35 %) and one patient had a progression-free survival of over 7 months. The most reported related toxicities were fatigue and dysgeusia. CP-4126 acts as a prodrug for gemcitabine when given orally, but because of the poor absorption, the rapid pre-systemic metabolism and consequently the amount of capsules the patient had to take (12 capsules at the highest dose), the study was terminated before the RD had been determined. Due to these findings, the development of oral CP-4126 was stopped and further research is focusing on the i.v. variant.
Ethical standards and conflict of interest
The study protocol was approved by the local Medical Ethics Committee and all patients had to give written informed consent. The ClinicalTrials.gov identifier is NCT00778128. W.Rasch, T. Bergeland and P.-A. Hals are employed at Clavis Pharma. W. Rasch owns stock in Clavis Pharma. The other authors declare that they have no conflict of interest.
References
Noble S, Goa KL (1997) Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs 54(3):447–472
Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE (1998) Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58(19):4349–4357
Bergman AM, Pinedo HM, Peters GJ (2002) Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug Resist Updat 5(1):19–33
Burke T, Lee S, Ferguson PJ, Hammond JR (1998) Interaction of 2′,2′-difluorodeoxycytidine (gemcitabine) and formycin B with the Na + -dependent and -independent nucleoside transporters of Ehrlich ascites tumor cells. J Pharmacol Exp Ther 286(3):1333–1340
Mini E, Nobili S, Caciagli B, Landini I, Mazzei T (2006) Cellular pharmacology of gemcitabine. Ann Oncol 17(Suppl 5):7–12
Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W (1990) Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2′,2′-difluorodeoxycytidine. Mol Pharmacol 38(4):567–572
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W (1991) Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res 51(22):6110–6117
Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W (1992) Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of self-potentiation. Cancer Res 52(3):533–539
Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW (2006) Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab Dispos 20(6):849–855
Plunkett W, Huang P, Searcy CE, Gandhi V (1996) Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol 23(5 Suppl 10):3–15
Banna GL, Collovà E, Gebbia V, Lipari H, Giuffrida P, Cavallaro S, Condorelli R, Buscarino C, Tralongo P, Ferraù F (2010) Anticancer oral therapy: emerging related issues. Cancer Treat Rev 36(8):595–605
Kerbel RS, Kamen B (2004) The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 4(6):423–436
André N, Padovani L, Pasquier E (2011) Metronomic scheduling of anticancer treatment: the next generation of multitarget therapy? Future Oncol 7(3):385–394
Cham KKY, Baker JHE, Takhar KS et al (2010) Metronomic gemcitabine suppresses tumour growth, improves perfusion, and reduces hypoxia in human pancreatic ductal adenocarcinoma. Br J Cancer 103(1):52–60
Laquente B, Lacasa C, Ginestà MM, Casanovas O, Figueras A, Galán M, Ribas IG, Germà JR, Capellà G, Viñals F (2008) Antiangiogenic effect of gemcitabine following metronomic administration in a pancreas cancer model. Mol Cancer Ther 7(3):638–647
Veltkamp SA, Jansen RS, Callies S et al (2008) Oral administration of gemcitabine in patients with refractory tumors: a clinical and pharmacologic study. Clin Cancer Res 14(11):3477–3486
Bender DM, Bao J, Dantzig AH et al (2009) Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J Med Chem 52(22):6958–6961
Koolen SLW, Witteveen PO, Jansen RS et al (2011) Phase I study of Oral gemcitabine prodrug (LY2334737) alone and in combination with erlotinib in patients with advanced solid tumors. Clin Cancer Res 17(18):6071–6082
Adema AD, Bijnsdorp IV, Sandvold ML, Verheul HM, Peters GJ (2009) Innovations and opportunities to improve conventional (deoxy)nucleoside and fluoropyrimidine analogs in cancer. Curr Med Chem 16(35):4632–4643
Bergman AM, Adema AD, Balzarini J et al (2011) Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs 29(3):456–466
Adema AD, Smid K, Losekoot N, Honeywell RJ, Verheul HM, Myhren F, Sandvold ML, Peters GJ (2012) Metabolism and accumulation of the lipophilic deoxynucleoside analogs elacytarabine and CP-4126. Invest New Drugs 30(5):1908–1916
Eisenhauer E, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Lilly E Gemzar 200 mg powder for solution for infusion, Gemzar 1000 mg powder for solution for infusion. Summary of Product Characteristics (SPC)
Fukami T, Yokoi T (2012) The emerging role of human esterases. Drug Metab Pharmacokinet 27(5):466–477
Abbruzzese JL, Grunewald R, Weeks EA, D G, Adams T, Nowak B, Mineishi S, Tarassoff P, Satterlee W, Raber MN (1991) A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J Clin Oncol 9(3):491–498
Peters GJ, Clavel M, Noordhuis P, Geyssen GJ, Laan AC, Guastalla J, Edzes HT, Vermorken JB (2007) Clinical phase I and pharmacology study of gemcitabine (2′, 2′-difluorodeoxycytidine) administered in a two-weekly schedule. J Chemother 19(2):212–221
Stuurman FE, Lolkema MPJ, Soetekouw PMMB, Rosing H, Mergui-Roelvink M, Beijnen JH, Voest EE, Van Tinteren H, Huitema ADR, Schellens JHM (2012) 581 an adaptive, 2-stage, phase 1 comparative pharmacokinetic and cardiac safety study of two intravenous formulations of CO-101 in patients with advanced solid tumors. Eur J Cancer Supp 48(supp 6):178
Veltkamp S, Pluim D, Van Eijndhoven MAJ, Bolijn MJ, Ong FHG, Govindarajan R, Unadkat JD, Beijnen JH, Schellens JHM (2008) New insights into the pharmacology and cytotoxicity of gemcitabine and 2′,2′-difluorodeoxyuridine. Mol Cancer Ther 7(8):2415–2425
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stuurman, F.E., Voest, E.E., Awada, A. et al. Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Invest New Drugs 31, 959–966 (2013). https://doi.org/10.1007/s10637-013-9925-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9925-z