
Summary
Purpose To determine the maximum tolerated dose (MTD), safety, pharmacokinetics, pharmacodynamics, and preliminary evidence of antitumor activity of the PI3K/mTOR inhibitor PF-04691502, administered orally once daily. Methods Escalating doses of PF-04691502 were administered to 23 patients with advanced solid tumors in sequential cohorts across the following dose levels: 2 mg, 4 mg, 8 mg, and 11 mg. 14 additional patients were enrolled in an expansion cohort at the MTD to ensure at least five matched pre- and post-treatment biopsies for biomarkers of PI3K activity. Results The MTD of PF-04691502 was 8 mg orally once daily. There were three dose-limiting toxicities: one grade 3 fatigue at 8 mg, one grade 3 rash at 11 mg, and one intolerable grade 2 fatigue at 11 mg. Among 37 patients enrolled, treatment-related adverse events included fatigue, decreased appetite, nausea, hyperglycemia, rash, and vomiting. Across all dose levels, average steady-state plasma PF-04691502 concentrations approximated or exceeded the target concentration of 16.2 ng/mL required for ≥75 % tumor growth inhibition in preclinical models. PF-04691502 resulted in increased mean fasting serum glucose, insulin, and c-peptide levels, and produced partial blockade of PI3K signalling in five paired tumor biopsies, as demonstrated by reductions in phosphorylated Akt, FKHR/FKHRL1, and STAT3. No objective anti-tumor responses were observed. Conclusions Daily oral administration of PF-04691502 was tolerable at 8 mg orally once daily, with a safety profile similar to other PI3K/mTOR inhibitors. PF-04691502 demonstrated PI3K pathway inhibition by changing glucose homeostasis, and by decreasing phosphorylation of downstream molecules in tumor tissue.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The PI3K/Akt/mTOR pathway is one of the principal signalling mechanisms regulating cell survival, growth, proliferation, metabolism, and motility [1–3]. Central to this pathway are the class 1 phosphatidylinositol 3-kinases (PI3Ks): heterodimers that consist of a catalytic subunit (p110α, p110β, or p110δ for class 1A, and p110γ for class 1B PI3Ks) and a regulatory subunit [1–3]. Class 1 PI3Ks, which are the PI3Ks most closely implicated in cancer, are activated through binding of the regulatory subunit by receptor tyrosine kinases (class 1A) or G-protein coupled receptors (class 1B) [1–4], or through direct activation of the catalytic subunit by Ras [5]. Following activation, the PI3K catalytic subunit phosphorylates phosphatidylinositol (4,5) biphosphate (PIP2) to phosphatidylinositol (3, 4,5) triphosphate (PIP3) in a reaction that is counter-balanced by phosphatase and tensin homolog (PTEN) [1–3]. PIP3 in turn recruits protein kinase B (Akt) to the plasma membrane, where it is co-activated through phosphorylation by phosphatidylinositol-dependent kinase 1 (PDK1) at T308 and the rictor-mTOR complex (mTORC2) at S473 [1–3]. Activated Akt phosphorylates multiple downstream targets, including hamartin/tuberin (TSC1/2); this latter action releases Ras homolog enriched in brain (Rheb) inhibition and thereby allows activation of mTOR through association with raptor and other mTORC1 complex members, culminating in cell growth and protein synthesis [1–3].
In malignancy, perturbation of the PI3K/Akt/mTOR pathway can occur through various mechanisms, including amplification or mutational activation of genes encoding receptor tyrosine kinases, Ras, and/or the p110α catalytic subunit of PI3K [PIK3CA], and inactivation of the tumor suppressor gene, PTEN [1–3]. The PI3K/Akt/mTOR pathway has long been a target for anticancer drug development, since it is implicated in tumor progression and development of resistance to anticancer drugs [1, 2]. The first PI3K/Akt/mTOR inhibitors were rapamycin analogs (e.g. everolimus, temsirolimus), which allosterically inhibit the raptor-mTOR complex (mTORC1) [6]. The limited clinical efficacy of these agents may be due to their inability to inhibit mTORC2, and the abrogation of negative feedback loops caused by inhibition of mTORC1 alone. The structural similarity between the catalytic domains of the p110 subunit of PI3K and mTOR provides the opportunity for targeting the PI3K/Akt/mTOR pathway at multiple sites with a single pharmacologic agent. Dual PI3K/mTOR inhibitors have the theoretical advantage of offering more complete suppression of the PI3K/Akt/mTOR pathway by inhibiting all catalytic isoforms of PI3K as well as mTORC1 and mTORC2 [7].
PF-04691502 is an orally active, ATP-competitive, dual inhibitor of PI3K and mTOR [8]. In vitro, PF-04691502 produces potent inhibition of recombinant class I PI3K (Ki = 1.2 to 2.1 nM) and mTOR (Ki = 16 nM) kinase activity, Akt phosphorylation (IC50 = 3.8 to 47 nM), and proliferation (IC50 = 179 to 313 nM) of PIK3CA-mutant and PTEN-null cancer cell lines [8]. In vivo, PF-04691502 inhibits tumor growth in glioblastoma (PTEN null U87), ovarian (PIK3CA mutant SKOV3), and erlotinib/gefitinib-resistant non-small cell lung cancer (PIK3CA mutant NCI-H1975 & NCI-H460, PTEN-null NCI-H1650, and KRAS mutant A549) xenograft models [8]. This first-in-human phase I clinical trial of PF-04691502 was designed to assess the drug’s safety and tolerability and to identify the maximum tolerated dose using a continuous daily dosing regimen. Secondary objectives were to characterize the single- and multiple-dose pharmacokinetics of PF-04691502, to determine its effects on PI3K-related pathways and markers of glucose metabolism, and to seek preliminary evidence of antitumor activity.
Patients and methods
Patient selection
Patients ≥18 years of age with histologically or cytologically confirmed malignant solid tumors for which there was no standard treatment, or which were unresponsive to treatment, were eligible for study inclusion. In addition, patients were required to have an Eastern Cooperative Oncology Group performance status 0 to 1; adequate glycemic control, including no previous diagnosis of diabetes mellitus and glycosylated hemoglobin (HbA1c) <7 %; and adequate bone marrow, renal, hepatic, and cardiac function. Patients were excluded if they had known, active brain metastasis; had received prior treatment with a PI3K- and/or mTOR-targeted agent; had significant cardiovascular disease, gastrointestinal abnormalities or known pulmonary impairment; or had current or anticipated use of drugs known to be potent CYP3A4 inhibitors or inducers.
Study design and treatment
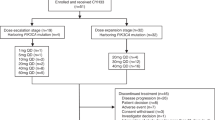
This Phase I, open-label, dose-escalation study employed a standard 3 + 3 method, whereby sequential cohorts of 3 to 6 patients were assigned progressively higher doses of PF-04691502 monotherapy, administered once daily in continuous 21-day treatment cycles. PF-04691502 was supplied as 1 mg and 5 mg tablets (Pfizer Inc., New York, NY) and was administered with ≥180 ml water in the fasted state (food was withheld for 2 h pre- and post-dosing). The first treatment cycle was preceded by a ≥7-day lead-in period to evaluate the single-dose pharmacokinetics of PF-04691502. Dose escalation proceeded initially in 100 % increments from a starting dose of 2 mg (based on an estimation of the STD10 [severely toxic dose in 10 % of animals] in rodent toxicology studies) until one patient in the dose cohort experienced a dose-limiting toxicity (DLT) and/or two patients experienced drug-related grade ≥2 toxicities during the first treatment cycle. Thereafter, dose escalation continued in ≤40 % increments, adjusted to accommodate the available tablet strengths, until DLT was observed in at least two of the three to six patients in the dose cohort. Intra-subject dose escalation was not permitted. The maximum tolerated dose (MTD) was defined as the highest dose associated with occurrence of DLT in <33 % of patients. DLT was defined as any one of the following events, as classified by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0, occurring during the first treatment cycle and attributable to PF-04691502: grade 4 neutropenia for ≥7 days; febrile neutropenia; grade 4 thrombocytopenia, or grade 3 thrombocytopenia with bleeding; grade 3 or 4 nausea, vomiting, or diarrhea despite medical intervention; other clinically significant grade ≥3 non-hematologic toxicity; or persistent, intolerable, drug-related toxicity that delays scheduled retreatment for >14 days. An expansion cohort of patients with paired pre-/post-treatment tumor biopsies was subsequently enrolled at the MTD to determine the effects of PF-04691502 on PI3K and mTOR signalling.
The study was approved by the institutional review board or independent ethics committee at each participating center, and was performed in accordance with the Declaration of Helsinki, Good Clinical Practice (GCP) guidelines and applicable local regulatory requirements and laws. All patients provided their written informed consent.
Assessments
Safety and tolerability
Comprehensive safety evaluations, including physical examination, vital signs, triplicate 12-lead ECGs, and clinical laboratory tests (hematology, blood chemistry, urinalysis), were performed at baseline and at regular intervals (days 8 and 15 of cycle 1, days 1 and 15 of cycle 2, day 1 of subsequent cycles) throughout the study, and on study completion. In addition, patients performed fasting home glucose monitoring 3-times weekly during cycle 1 and weekly during cycle 2. Adverse events were assessed for severity and relationship to treatment and were graded according to the NCI-CTCAE, version 4.0.
Pharmacokinetics
Following administration of a lead-in dose of PF-04691502 (4 to10 days prior to commencement of continuous daily dosing), serial blood samples were collected at 0 (pre-dose), 0.5, 1, 2, 4, 6, 8, 24, 48, 72 and 96 h post-dose for single-dose pharmacokinetic assessment. Steady-state pharmacokinetics were assessed on day 21 of the first treatment cycle, using blood samples collected at 0 (pre-dose), 0.5, 1, 2, 4, 6, 8 and 24 h post-dose. Plasma PF-04691502 concentrations were measured using a validated, sensitive and specific high-performance liquid chromatography tandem mass spectrometric (HPLC-MS/MS) technique, with a lower limit of quantification of 0.25 ng/mL. The following pharmacokinetic parameters were estimated by non-compartmental analysis using internally validated Pfizer software (eNCA, version 2.2.3, Groton, CT): maximum plasma concentration (Cmax), time to reach Cmax (tmax), area under the plasma concentration‒time curve from time zero to 24 h (the dosing interval; AUC0-24 h), and the terminal elimination half-life (t½). AUC values were calculated using the log-linear trapezoidal method, and t½ values were derived from linear regression of the log-linear concentration-time curve.
Pharmacodynamics
Archived (baseline) tumor biopsy samples were analyzed for evidence of PI3K pathway activation, including the presence of activating mutations in PIK3CA, PIK3CA amplification and PTEN protein loss. Hair follicles were collected at baseline, and at pre-dose and 2, 4, and 24 h post-dose on day 21 of the first treatment cycle for reverse phase protein microarray (RPMA) analysis of proteins involved in PI3K/mTOR signalling and mitosis, including pAKT (S473), pAkt (T308), pSTAT3 (Y705), Ki-67, and pPRAS40 (T246). In addition, fresh, paired tumor biopsies (expansion cohort only) were obtained at baseline and at approximately 4 h post-dose on day 21 of the first treatment cycle for RPMA analysis of biomarkers of PI3K activity, including pAkt (S473), pAkt (T308), pSTAT3 (Y705), and pFKHR (T24)/FKHRL1 (T32). The RPMA readout, a fluorescence signal, was normalized for cytokeratin (NFC). Fasting serum glucose, C-peptide, insulin, hemoglobin and HbA1c levels were measured using blood samples drawn at 1 h post-dose during cycle 1 (days 8 and 15), cycle 2 (days 1 and 15) and subsequent treatment cycles (day 1).
Antitumor activity
Baseline tumor assessment was performed within 28 days prior to initiation of PF-04691502 treatment, and restaging assessments were performed at 6-week intervals, prior to commencement of each odd-numbered treatment cycle. Objective tumor response was evaluated according to RECIST criteria, version 1.1 [9].
Statistical methods
The study sample size was determined empirically based on the occurrence of DLT in the dose-escalation cohort. The dose-expansion cohort was designed to ensure collection of at least five pairs of pre-/post-treatment tumor biopsies. Safety, efficacy, pharmacokinetic, and pharmacodynamics findings were summarized with descriptive statistics.
Results
Patient characteristics
A total of 37 patients (17 male, 20 female) were enrolled at three study centers in the United States and treated with PF-04691502. The study population was predominantly Caucasian (86.5 %), of mean age 59.6 (range 36 to 81) years, and the most common tumor types were colorectal and non-small cell lung cancer (Table 1). Overall, this was a heavily pre-treated population, with 81 % of patients having previously received ≥3 systemic treatment regimens.
Dose escalation and expansion
Patients were treated with PF-04691502 across 4 dose levels: 2 mg (n = 3), 4 mg (n = 3), 8 mg (n = 12) and 11 mg (n = 5) once daily in the dose-escalation phase; subsequently, a further 14 patients were treated with PF-04691502 at the 8 mg dose level in the dose-expansion phase of the study. Overall, treatment was administered for a median of 18.1 weeks (2 mg dose cohort), 6.1 weeks (4 mg dose cohort), 5.1 weeks (8 mg dose cohort) and 2.1 weeks (11 mg dose cohort), with individual treatment durations ranging from 0 (lead-in dose only) to 24 weeks.
Among the dose-escalation cohorts, six patients were not evaluable for DLT, due to progressive disease and/or patient withdrawal prior to completion of course 1. Three patients experienced dose-limiting toxicities during the first treatment cycle, as depicted in Table 2. One patient experienced grade 3 fatigue on day 16 at the 8 mg dose level, which resolved after temporary discontinuation of the study drug. Two patients experienced dose-limiting toxicity at the 11 mg dose level: one had grade 3 maculopapular rash, and the other had grade 2 fatigue. The 8 mg dose level was established as both the maximum tolerated dose and the recommended phase II dose.
Safety and tolerability
All 37 patients treated with PF-04691502 were included in the safety assessment. Treatment-related adverse events, presented by PF-04691502 dose level, that were reported in ≥5 % of patients are summarized in Table 3. Overall, the most frequent treatment-related adverse events in the study population were fatigue (40.5 %), decreased appetite (35.1 %), nausea (35.1 %), hyperglycemia (27.0 %), rash (27.0 %), vomiting (27.0 %), diarrhea (18.9 %) and mucosal inflammation (18.9 %).
There were no treatment interruptions or discontinuations for treatment-related adverse events at the first 2 dose levels. Four patients permanently discontinued PF-04691502 at the 8 mg dose level due to treatment-related grade 3 colitis (n = 1), rash (n = 1), headache (n = 1), and dyspnea (n = 1), and two patients discontinued at the 11 mg dose level due to dose-limiting toxicities. Twelve patients required temporary dose interruption at the 8 mg and 11 mg dose levels due to treatment-related adverse events, most commonly for grade 2 to 3 hyperglycemia (n = 6). Blood glucose levels normalized following treatment withdrawal, and PF-04691502 dosing was successfully resumed after initiation of oral hypoglycemic therapy. Other treatment-related adverse events resulting in temporary dose interruption included fatigue, diarrhea, and rash, either alone or in combination.
Overall, 14 of 37 (37.8 %) patients experienced treatment-related dermatologic adverse events, most commonly presenting as a maculo-papular rash that was manageable with PF-04691502 interruption and/or supportive care. However, the rash could also be severe: one patient at 11 mg had dose-limiting rash as outlined above, and another at 11 mg was hospitalized for rash within 2 weeks of withdrawal from study due to progressive disease.
There were three serious pulmonary adverse events possibly related to PF-04691502. One patient came off study for progressive lung metastases after six cycles at 8 mg, and was admitted to hospital with acute respiratory distress syndrome 1 week later. Another patient, who came off study for progressive lung metastases after nearly three cycles at 8 mg, developed dyspnea within 2 weeks of the last study drug and was found to have pneumonitis. A third patient required hospitalization for grade 3 dyspnea during cycle 8 at 8 mg, and improved after PF-04691502 was permanently discontinued.
PF-04691502 had no clear dose-related effect on QTc interval. Five patients with normal baseline QTc intervals developed grade 1 or 2 QTc prolongation at the 8 mg dose level, with one patient showing a ≥60 msec increase from baseline.
Pharmacokinetics
PF-04691502 was rapidly absorbed after oral administration, with a median time to peak plasma drug concentration of approximately 2 to 4 h, and eliminated in a biphasic manner, with a terminal elimination half-life of approximately 9 to 12 h. Median plasma PF-04691502 concentration-time profiles after single-dose administration and at steady state (Day 21 of once daily dosing) are depicted in Fig. 1. After both single and repeated dose administration, peak and total plasma PF-04691502 exposure increased in a dose-related, although somewhat less than dose-proportional, manner over the 2 to 11 mg dose range, as indicated by mean Cmax and AUC0-24h values (Table 4). Intersubject variability in Cmax and AUC0-24h values, based on geometric CV%, was between approximately 15 to 40 % and 5 to 45 % on single- and repeated-dose administration, respectively. Moderate accumulation of PF-04691502 was noted at steady-state, with the mean accumulation ratio ranging from 1.4 to 1.7 based on AUC0-24h values and from 1.2 to 1.6 based on Cmax values. At steady-state, average plasma PF-04691502 concentrations over the 24-h dosing interval (15.5, 40.3, 53.6 and 42.5 ng/ml with 2, 4, 8 and 11 mg doses, respectively) approximated to or exceeded the target concentration of 16.2 ng/ml (38 nM) required for simultaneous suppression of pAKT (≥50 % suppression) in cell assays and ≥75 % tumor growth inhibition in preclinical xenograft models [8].

Median plasma PF-04691502 concentration-time profiles following a single-dose and b multiple-dose oral administration of PF-04691502 at doses of 2, 4, 8 and 11 mg once daily
Pharmacodynamics
Among archived tumor specimens tested for evidence of PI3K pathway activation, 9 of 18 (50 %) were PTEN-low, 2 of 17 (11.8 %) showed PIK3CA amplification, and 3 of 17 (17.6 %) had a detectable PIK3CA mutation [the mutations found were R93W (exon 1), E545K (exon 9), and M1043I (exon 20)]. Correlation of biomarkers with response status suggested that patients whose tumors were deficient in PTEN were more likely to experience disease progression with PF-04691502 than those with PTEN-normal tumors (P = 0.0498). There was no significant difference in PIK3CA amplification or mutation status between patients with stable disease and those with disease progression. Analysis of hair follicles showed reductions in pAkt (S473), pPRAS40 (T246), and pSTAT3 (Y705) at 4 h post-dose on Day 21 of treatment cycle 1 relative to baseline, but there was a rebound in signalling that was maintained, even after a subsequent dose.
The expansion cohort provided five paired tumor biopsies for analysis of biomarkers of PI3K pathway activity, although for pAkt(T308), there were only four evaluable pairs. PF-04691502 produced a partial blockade of tumor PI3K signalling, as reflected in the mean (± standard deviation) absolute change in fluorescence (NFC) signal from baseline to Day 21 in pAkt (S473) of -72.6 (±82.26); pAkt (T308) of -48.6 (±40.37); pFKHR (T24)/FKHRL1 (T32) of -81.5 (±47.70); and pSTAT3 (Y705) of -112.2 (±78.14).
Analysis of markers of glucose metabolism showed increases from baseline in mean fasting serum glucose (16 to 23 %), insulin (approximately 2- to 7-fold) and c-peptide (approximately 2-fold) levels after 8 days of PF-04691502 treatment across the dose levels. In total, 18 of the 26 patients treated with PF-04691502 at the 8 mg dose level showed an increase from baseline in fasting serum glucose level at this time point. Modest reductions in mean serum haemoglobin (2 to 8 %) and increases in the proportion of glycosylated hemoglobin (HbA1c) to total hemoglobin (7 to 16 %) from baseline to the end of PF-04691502 treatment were recorded.
Antitumor activity
No objective radiographic responses were obtained among the 36 patients who were evaluable for treatment efficacy. A best response of stable disease was recorded in 12 (33.3 %) patients, while 13 (36.1 %) patients showed objective evidence of disease progression, 9 (25 %) had indeterminate response, and 2 (5.6 %) had early death.
Discussion
This phase I study demonstrated that PF-04691502 was well tolerated at doses of up to 8 mg orally once daily, and 8 mg was determined to be the MTD. The most frequent toxicities observed with PF-04691502 - fatigue, decreased appetite, nausea, hyperglycemia, rash, and vomiting - are commonly associated with PI3K/Akt/mTOR pathway inhibitors [2, 7, 10], and were generally tolerable and/or manageable with supportive care. Pulmonary effects have previously been noted with PI3K/mTOR inhibitors [2, 7], and the possibility that PF-04691502 may have contributed to development of dyspnea in this study cannot be excluded. However, concurrent progressive pulmonary metastasis was a confounding factor.
Pharmacokinetic evaluation indicated an apparent elimination half-life for PF-04691502 ranging from 10.9 to 13.8 h across all dose levels, which would support once daily administration. At doses of 4 mg and higher, steady-state mean plasma PF-04691502 concentrations over the 24-h dose interval exceeded target concentrations for inhibition of the PI3K/Akt/mTOR pathway in vitro and slowing of tumor growth in pre-clinical models.
A number of pharmacodynamic and potential biomarkers for patient tailoring were explored in analyses of archival tumor tissue, paired tumor biopsies, paired hair follicle samples, and blood. PTEN protein and PIK3CA were examined in archival tumor tissues as possible predictive biomarkers, but similar to the experience with other pan-PI3K inhibitors, this analysis was inconclusive [10]. Studies involving the pharmacodynamic biomarkers were more informative, with post-treatment tumor biopsies and hair follicle samples demonstrating inhibition of PI3K signalling. The observed increases in blood glucose, c-peptide, and insulin levels, which generally occurred by Day 8 of the first treatment cycle at the 8 mg dose level, are also consistent with PI3K/mTOR pathway inhibition, since PI3K mediates the metabolic effects of insulin [11, 12]. PI3K/mTOR inhibitors induce metabolic effects akin to a fasted metabolic state, with increased fatty acid oxidation and decreased glucose utilization, and have been associated with metabolic toxicities of hyperlipidemia and hyperglycemia [13]. Evidence from this study suggests that PF-04691502 blocks glucose transport and/or metabolism, leading to high glucose levels that are rapidly countered by increased proinsulin processing, with consequent c-peptide cleavage and insulin secretion.
Despite this continuous drug exposure and evidence of down-regulation of PI3K signaling, none of the 25 heavily pretreated patients evaluable for efficacy demonstrated an objective tumor response to PF-04691502. In contrast to PF-04691502, weekly intravenous administration of another pan-PI3K/mTOR inhibitor, PF-05212384, results in biologically-relevant serum drug concentrations for 5 out of 7 days, and has demonstrated objective tumor responses [14]. While the difference in clinical efficacy may be due to intrinsic features of the drugs themselves, it is conceivable that continuous inhibition of PI3K/mTOR at the levels demonstrated via the pharmacodynamic biomarkers may result in an adaptation favoring cell growth. For certain conditions harboring driver mutations, such as BCR-ABL in chronic myeloid leukemia, the continued inhibition of kinase activity provided by higher trough concentrations of targeted therapy results in more favourable clinical outcomes [15, 16]. In other malignancies, however, the requirement for continuous inhibition is being challenged by models of drug resistance [17]. For pan-PI3K/mTOR-targeted drugs, further studies are required to determine whether selective pressures are operative in cancer patients and, if so, whether these can be modulated by intermittent treatment.
As a class, pan-PI3K inhibitors generally show cytostatic rather than cytotoxic clinical activity, reflected in disease stabilization rather than tumor regression in patients with advanced solid tumors [10, 18]. In a breast cancer cell line variant with increased PI3K activation and high metastatic bone tropism (MDA-MB-231-1833), nanomolar concentrations of PF-04691502 did not impair cell proliferation in vitro; however, following implantation into mice, these PF-04691502-pretreated cells showed reduced ability to establish bone metastasis in vivo [24]. This suggests that there may be grounds for examining the activity of PI3K/MTOR inhibitors in earlier stages of disease, and/or in less heavily pre-treated patients.
A growing body of evidence suggests that PI3K/mTOR inhibitors may be more effective when used in combination with standard chemotherapy, hormonal therapy, radiation therapy, and/or newer targeted agents [18, 19]. The PI3K/mTOR and RAS/RAF/MEK pathways intersect at multiple points [20], and PF-04691502 appears to be more effective when used in combination with inhibitors of RAS/RAF/MEK signalling [21]. For example, in a genetically engineered murine model of ovarian cancer driven by epithelial KRAS G12D activation and PTEN deletion, single-agent PF-04691502 inhibited PI3K signalling, tumor glucose metabolism, and tumor growth, but failed to prevent tumor progression on continued administration [21]. This would suggest that these tumors are able to overcome their dependency on the PI3K/mTOR pathway through activation of signalling cascades downstream from KRAS G12D. In contrast to single-agent PF-04691502, the combination of PF-04691501 and the MEK inhibitor PD-0325901 resulted in pronounced and sustained tumor regression in the KRAS G12D;PTEN del model [21]. Similarly in KRAS G12D-LSL conditional genetically engineered mouse models, the inhibitory effect of PF-04691502 on hyperplastic, benign adenoma, and adenocarcinoma lung lesions was enhanced by sub-MTD doses of PD-0325901 [22]. Regarding combinations with traditional anti-cancer therapies, the addition of PF-04691502 to radiation delays tumor growth in a human wild-type P53 head and neck squamous cell carcinoma (HNSCC) xenograft model [23]. Taken together, these preclinical data suggest that the promise of PI3K/mTOR inhibitors for solid tumors may not be realized until the agents are studied in rational combinations, potentially in preselected patient populations.
In conclusion, PF-04695102 was well tolerated at an oral dose of 8 mg once daily, with a toxicity profile consistent with that of other PI3K/mTOR inhibitors. The pharmacokinetic profile supported once daily dosing, and pharmacodynamic studies in tumor tissue and hair follicles confirmed target PI3K inhibition. No objective tumor responses were observed with PF-04691502, whereas a concurrent first-in-human study of an alternate PI3K/mTOR inhibitor (PF-05212384) demonstrated anti-tumor activity [25]. Further development of PF-04691502 is on hold, due to the lack of objective responses with the drug, and a higher than expected incidence of serious rash in a subsequent clinical trial of PF-04691502 plus letrozole in breast cancer [26].
References
Courtney KD, Corcoran RB, Engelman JA (2010) The PI3K pathway as drug target in human cancer. J Clin Oncol 28:1075–1083
Ogita S, Lorusso P (2011) Targeting phosphatidylinositol 3 kinase (PI3K)-Akt beyond rapalogs. Target Oncol 6:103–117
Ihle NT, Powis G (2009) Take your PIK: phosphatidylinositol 3-kinase inhibitors race through the clinic and toward cancer therapy. Mol Cancer Ther 8:1–9
Arcaro A, Guerreiro AS (2007) The phosphoinositide 3-kinase pathway in human cancer: genetic alterations and therapeutic implications. Curr Genomics 8:271–306
Rodriguez-Viciana P, Warne PH, Dhand R et al (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370:527–532
Abraham RT, Eng CH (2008) Mammalian target of rapamycin as a therapeutic target in oncology. Expert Opin Ther Targets 12:209–222
Markman B, Dienstmann R, Tabernero J (2010) Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget 1:530–543
Yuan J, Mehta PP, Yin MJ et al (2011) PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther 10:2189–2199
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Rodon J, Dienstmann R, Serra V et al (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10:143–153
Knight ZA, Gonzalez B, Feldman ME et al (2006) A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125:733–747
Foukas LC, Claret M, Pearce W et al (2006) Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441:366–370
Busaidy NL, Farooki A, Dowlati A et al (2012) Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol 30:2919–2928
Millham R, Houk B, Borzillo G, et al (2011) First-in-patient study of PF-05212384, a small molecule intravenous dual inhibitor of PI3K and mTOR in patients with advanced cancer: update on safety, efficacy, and pharmacology. Mol Cancer Ther 10:Abst. A167
Larson RA, Druker BJ, Guilhot F et al (2008) Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood 111:4022–4028
Picard S, Titier K, Etienne G et al (2007) Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood 109:3496–3499
Das Thakur M, Salangsang F, Landman AS et al (2013) Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494:251–255
Britten CD (2013) PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 71:1395–1409
Burris HA 3rd (2013) Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol 71:829–842
Mendoza MC, Er EE, Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36:320–328
Kinross KM, Brown DV, Kleinschmidt M et al (2011) In vivo activity of combined PI3K/mTOR and MEK inhibition in a Kras(G12D);Pten deletion mouse model of ovarian cancer. Mol Cancer Ther 10:1440–1449
Simmons BH, Lee JH, Lalwani K et al (2012) Combination of a MEK inhibitor at sub-MTD with a PI3K/mTOR inhibitor significantly suppresses growth of lung adenocarcinoma tumors in Kras(G12D-LSL) mice. Cancer Chemother Pharmacol 70:213–220
Herzog A, Bian Y, Vander Broek R et al (2013) PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res 19:3808–3819
Wander SA, Zhao D, Besser AH et al (2013) PI3K/mTOR inhibition can impair tumor invasion and metastasis in vivo despite a lack of antiproliferative action in vitro: implications for targeted therapy. Breast Cancer Res Treat 138:369–381
Mallon R, Feldberg LR, Lucas J et al (2011) Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res 17:3193–3203
Canon JL, Bergh J, Saura C et al (2012) Phase Ib/II study of an oral PI3K/mTOR inhibitor plus letrozole compared with letrozole (L) in pre-operative setting in patients with Estrogen Receptor-positive, HER2-negative early breast cancer (BC): Phase Ib preliminary data. Cancer Res 72:572S, Abst. OT2-3-02
Ethical standards
This clinical trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice (GCP) guidelines and applicable local regulatory requirements and laws.
Conflict of interest
CDB has received clinical research support and consulting fees from Pfizer Inc. AAA, ZAW, and PMLR have received clinical research support. RM, BEH, GB, and KP were employees of Pfizer Inc. at the time of this study, and may hold stock options in the company.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Britten, C.D., Adjei, A.A., Millham, R. et al. Phase I study of PF-04691502, a small-molecule, oral, dual inhibitor of PI3K and mTOR, in patients with advanced cancer. Invest New Drugs 32, 510–517 (2014). https://doi.org/10.1007/s10637-013-0062-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-0062-5