
Summary
The aim of this study was to determine the maximum tolerated dose, dose-limiting toxicities, and pharmacokinetic profile of E7107 in patients with advanced solid tumors. Patients in this phase I, open-label, single-arm, dose-escalation study had metastatic or locally advanced solid tumors and received E7107 as a 30-minute intravenous infusion at doses of 0.6, 1.2, 1.8, 2.4, 3.2, 4.3, and 5.7 mg/m2. Twenty-six patients were enrolled in the study. At 5.7 mg/m2, two patients experienced dose-limiting toxicities including diarrhea, vomiting, dehydration, and myocardial infarction on Days 1–3 following E7107 administration. Three additional patients were recruited at the lower dose and all six patients tolerated E7107 4.3 mg/m2 with no dose-limiting toxicities. The maximum tolerated dose of E7107 was therefore 4.3 mg/m2. The most common drug-related adverse events were nausea, vomiting, and diarrhea. Vision loss was experienced by two patients at Cycles 2 and 7, each patient receiving 3.2 mg/m2 and 4.3 mg/m2, respectively. This resulted in the study being put on clinical hold. Pharmacokinetic analysis showed that E7107 was rapidly distributed with a moderate elimination half-life (6–13 h) and high clearance. Exposure to E7107 was dose-related. The best tumor response was stable disease in eight patients. E7107 is a unique first-in-class molecule. The incidence of two cases of vision loss probably related to E7107 led to study discontinuation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
E7107, a semi-synthetic urethane derivative of the natural fungal product pladienolide B, modulates protein expression via inhibition of spliceosome assembly and exhibits distinct broad-spectrum antitumor activity in preclinical studies. This report describes one of two first-in-human phase I dose-escalation studies of E7107 in patients with metastatic or locally advanced solid tumors.
E7107 has been shown to broadly inhibit precursor messenger ribonucleic acid (pre-mRNA) splicing by binding to spliceosome-associated protein-130, inhibiting spliceosome assembly [1, 2]. The results of this modulation of pre-mRNA processing may include both quantitative and qualitative effects on cellular protein expression. More specifically, inhibition may result in a global reduction in protein synthesis or a change in the relative abundance of various proteins. In addition, potential influences on alternative splicing patterns for certain pre-mRNA transcripts may result in a change in the expression of different alternatively spliced isoforms of important proteins. Consistent with the anti-tumorigenic effects of E7107, there is now increasing evidence that alternative splicing and alternatively spliced protein products are involved in cancer development [3].
How E7107-related alterations of pre-mRNA splicing might result in modulation of gene expression and exert effects on downstream events is being assessed. Flow cytometric analyses have demonstrated that E7107 induces cell-cycle arrest at both the G1 and G2/M phases. In addition, this agent exhibits selectivity against human tumor cell lines with functional loss of retinoblastoma (Rb) tumor-suppression protein and increased expression of p16INK4A and cyclin E [4].
E7107 inhibits the growth of tumor cell lines in vitro with nanomolar (1–20 nM) potency [5] and induces tumor regression in most human xenograft models tested, including lung, breast, and colon tumors [4, 6]. E7107 has a distinct broad-spectrum antitumor profile in vitro, including activity in a panel of cell lines that are resistant to classic cytotoxic drugs [7].
The primary objective of this phase I study was to determine the maximum tolerated dose (MTD) and pharmacokinetic (PK) profile of E7107 when administered as a 30-min infusion on Days 1 and 8 every 21 days. Secondary objectives included identifying dose-limiting toxicities (DLTs), exploring safety and tolerability, assessing preliminary indicators of efficacy, and identifying the potential for measuring changes in gene splicing and mRNA in peripheral blood cells as a pharmacodynamic biomarker for E7107.
Patients and methods
Study design and treatment
This was an open-label, single-arm, dose-escalation study (Study E7107-A001-101; Trial registration ID: NCT00499499) of E7107 administered as a 30-min intravenous infusion on Days 1 and 8 of each 21-day cycle in patients with solid tumors. The MTD was established by determining the occurrence of DLTs during the first 3 weeks of therapy (Cycle 1). Patients were treated at multiple dose levels, starting at 0.6 mg/m2. In this study, an accelerated design was used for dose escalation, with dose increases of 100 % until a grade 2 or higher adverse event (AE) occurred, at which time dosing was increased by 50 %, based on previously described designs [8, 9]. Patients were recruited to the next higher dose if one patient at the lower dose completed Cycle 1 with no drug-related toxicity greater than grade 1 (except alopecia, lymphocytopenia, or anemia). Initially, three patients were entered at each dose level; if one of the three patients experienced a DLT then three additional patients were entered into this treatment group. Dose escalation was permitted if there were no further DLTs at this dose. Intra-patient dose escalation was not permitted until the MTD was reached.
Patients were assessed at baseline (within 2 weeks before the start of treatment) and at Days 1, 2, 3, 8, 9, 10, and 15 of Cycle 1. Further assessments were performed at Days 1, 8, and 15 of any additional cycles. A final assessment was performed within 30 days of the last E7107 infusion. Follow-up assessments were performed every 3 months until death or until the patient withdrew consent.
The study was conducted in accordance with the Declaration of Helsinki. The protocol was approved by local institutional review boards and ethics committees, and patients provided written informed consent prior to the study.
Patients
The study recruited patients aged ≥18 years with metastatic or locally advanced solid tumors who had relapsed following treatment and for whom no other effective therapies were available. Key inclusion criteria included: no surgery or radiotherapy in the 4 weeks prior to study; discontinuation of prior chemotherapy and other anticancer therapy, excluding bisphosphonates at a steady dose level, at least 2 weeks prior to the study, provided there were no treatment-related acute toxicities identified; Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; and anticipated life expectancy >3 months.
Key exclusion criteria included: symptomatic or progressive brain tumors, or brain or leptomeningeal metastases requiring clinical intervention (unless patients had completed local therapy and corticosteroids had been discontinued at least 2 weeks before starting E7107 treatment); hemoglobin <9 g/dL; neutrophils <1.5 × 109/L; platelets <100 × 109/L; serum bilirubin >1.5 mg/dL; liver function tests (aspartate aminotransferase and alanine aminotransferase) >3 × upper limit of normal (ULN) (5 × ULN if liver metastases were present); serum creatinine >1.5 × ULN or creatinine clearance <40 mL/min; human immunodeficiency virus, active hepatitis B, active hepatitis C, or other severe or uncontrolled illness or infection; clinically significant cardiac impairment or unstable ischemic heart disease (including myocardial infarction [MI] within 6 months of study start); bleeding or thrombotic disorders, or the use of therapeutic dosages of anticoagulants; history of alcoholism, drug addiction, or any psychiatric or psychological condition; pregnancy or breastfeeding; prolongation of QT/QTc interval at screening; history of risk factors for torsades de pointes; concomitant use of medications that prolong QT/QTc interval; radiation therapy encompassing ≥25 % of bone marrow; history of hypersensitivity to pladienolide B or derivatives and excipients of the formulation; other significant comorbid conditions; organ allografts; and use of strong cytochrome P450 3A4 inhibitors 2 weeks prior to the study.
Assessments
Maximum tolerated dose
MTD was defined as the dose at which no more than one out of six patients experienced a DLT in Cycle 1. Only patients who completed Cycle 1 of treatment, and those who could not complete owing to toxicity, contributed to the determination of the MTD. DLTs were defined as: any grade 4 hematologic toxicity except neutropenia lasting <7 days and recovering to grade ≤2 without fever; neutropenia grade ≥3 complicated by fever or infection; thrombocytopenia grade ≥3 complicated by bleeding and requiring platelet or blood transfusions; non-hematologic toxicities grade ≥3 (except grade 3 nausea and vomiting uncontrolled by antiemetics, insomnia, obesity or weight gain, infertility, amenorrhea, galactorrhea, glucose intolerance due to dexamethasone use for antiemesis, asymptomatic hypercholesterolemia and hypertriglyceridemia, and non-hematologic toxicities grade ≥3 due to disease and disease progression); delayed recovery from drug-related toxicity, which delayed scheduled retreatment for >14 days; QTc interval >500 ms or 60 ms above baseline while on therapy; and toxicities that required dose reductions during Cycle 1.
Adverse events and other safety variables
AEs were recorded and monitored throughout the study, coded using the Medical Dictionary for Regulatory Activities (Version 10) and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; Version 3.0) or the three-point scale of mild, moderate, and severe, and by relationship to study medication (not related, possibly related, or probably related). AEs were presented by severity when the CTCAE grade was not collected. AEs were regarded as being treatment emergent (TEAEs) if they started on or after administration of the first dose of E7107 (but not later than 30 days after discontinuation), or if they increased in severity during treatment with E7107. Other parameters assessed included clinical laboratory measurements, vital signs, physical examinations, ECOG performance status, and electrocardiograms.
Pharmacokinetic evaluation
PK analyses of the parent compound and its active oxidized metabolite (M8; Eisai, data on file) were performed. Blood samples were collected immediately prior to E7107 infusion on Days 1 and 8 of Cycle 1. Patients were also sampled mid-infusion (5 and 15 min), at the end of the infusion, and at various time points post-infusion (5, 10, 15, and 30 min, and 1, 2, 3, 4, 6, 8, 12, 16, 24, and 48 h). Plasma samples were analyzed by means of a validated liquid chromatography-tandem mass spectrometry method. The limit of quantification (LOQ) for E7107 in plasma was 0.01 ng/mL, and the LOQ for M8 was 0.03 ng/mL. PK parameters were derived from model-independent analysis using the WinNonlin program (Version 5.1.1; Pharsight Corporation, CA). Urine samples were collected on Days 1 and 8 of Cycle 1 and for 48 h in the following intervals: 0–8 h, 8–24 h, and 24–48 h.
PK parameters measured included: time to peak concentration (t max); peak plasma concentration (C max); terminal phase elimination half-life (t ½); area under the plasma concentration-time curve extrapolated to time (AUC0–t ); area under the plasma concentration-time curve extrapolated to infinity (AUC0–∞); total body clearance from plasma (CL); volume of distribution at steady rate (V ss); and the fraction of dose excreted in urine (F e).
Pharmacodynamic analysis
Effects on gene splicing in peripheral blood mononuclear cells (PBMCs) in response to E7107 were monitored as a pharmacodynamic measure, as a function of both time and exposure to drug (recorded as total dose, AUC, or C max). Alternative splicing was monitored using the Affymetrix GeneChip® Human Exon 1.0 ST Array. This microarray can be used to assess the level of mRNA expression for all known human exons and many introns, and has 5,362,207 features used to interrogate one million exon clusters with over 1.4 million probe sets [10]. The total number of expressed introns was plotted against the time and dose relationship for altered expression of unspliced introns.
Efficacy
Baseline tumor assessments were carried out within 4 weeks of starting the study and included clinical examination and photography, computed tomography, or magnetic resonance imaging scans. Subsequent evaluations were performed every two cycles thereafter. Tumor assessments included overall visit response (complete response, partial response, stable disease [SD], and progressive disease [PD]) and best response according to Response Evaluation Criteria in Solid Tumors (RECIST) [11]. There was no formal statistical analysis of efficacy data.
Statistical analysis
The number of patients planned for enrollment in this study was estimated to be 25–40, based on the toxicities observed and the number of doses investigated. The safety population was used for analysis of safety and efficacy data and included all patients who received any dose of E7107. The PK population included all patients who had at least one evaluable post-dose PK sample.
Results
Patients
Twenty-six patients were enrolled in the study and all were included in both the safety and PK populations. Baseline demographics and clinical characteristics for all patients are shown in Table 1. Of the 26 patients, 25 (96.2 %) had previous chemotherapy and 19 (73.1 %) had radiotherapy. Study discontinuations were due to PD (16 patients; 61.5 %), AEs (4 patients; 15.4 %), clinical progression (3 patients; 11.5 %), physician’s decision to stop treatment (1 patient; 3.8 %), and other reasons not specified (1 patient; 3.8 %). One patient (3.8 %) was lost to follow-up. Two patients died during the study; one due to serious AEs (SAEs; grade 4 renal failure, respiratory failure, and septic shock) and the other due to disease progression. The deaths were considered unrelated to the study drug.
Study-drug exposure, MTD, and DLTs
The number of patients receiving each dose of E7107 and the number of cycles of each dose are summarized in Table 2. At E7107 5.7 mg/m2, two patients experienced serious TEAEs with DLT; one patient experienced DLTs of diarrhea (grade 2) and MI (grade 4), which required a treatment delay of 21 days and a dose reduction to E7107 4.3 mg/m2, and one patient experienced DLTs of diarrhea, vomiting, and dehydration (all a minimum of grade 3), which led to study withdrawal during Cycle 1. As a consequence of this, three additional patients were evaluated at a dose level of 4.3 mg/m2. All six patients tolerated this dose without a DLT. Therefore, the MTD was determined to be 4.3 mg/m2.
Safety
TEAEs were experienced by all 26 patients in this study. Of these, 24 patients (92.3 %) had TEAEs that were considered related to treatment. The most common treatment-related TEAEs (reported by ≥10 % of patients) were: nausea (76.9 %); vomiting (38.5 %); diarrhea (34.6 %); fatigue (30.8 %); anorexia (15.4 %); and peripheral neuropathy (11.5 %). Overall, nine patients (34.6 %) experienced TEAEs that were grade ≥3. Grade 4 TEAEs were reported by one patient receiving E7107 3.2 mg/m2 and by two patients receiving E7107 5.7 mg/m2; no grade 5 TEAE was reported by any patient in the study. TEAEs grade ≥3 considered to be treatment-related occurred in three patients (11.5 %; Table 3).
Nine patients (34.6 %) experienced SAEs during the study. The only SAEs reported by ≥2 patients included diarrhea (grade 3), increased blood creatinine (grade 2), and dehydration (grades 2 and 3). Serious TEAEs considered drug-related were reported by five patients (19.2 %) and two patients (7.7 %) experienced SAEs that were considered DLTs. Diarrhea was the only drug-related SAE reported by >5 % of patients. Five patients (19.2 %) discontinued the study as a consequence of TEAEs. Of these, four patients had SAEs related to study drug (vision loss [n = 2]; diarrhea, vomiting, and nausea [n = 1]; hypotension [n = 1]). One patient discontinued because of jugular vein thrombosis not related to E7107.
There were no clinically meaningful changes in hematology, chemistry, urinalysis, or vital signs during the study. Patients’ ECOG performance status did not worsen during the study. Two patients experienced an increase in QT/QTc value >60 ms from baseline, one on Day 8 of Cycle 2 and the other on Day 1 of Cycle 1. One patient experienced grade 1 QT prolongation reported as a TEAE, which was considered possibly related to study drug and resolved with no action taken.
Visual toxicity
One patient receiving E7107 3.2 mg/m2 and one patient receiving E7107 4.3 mg/m2 experienced vision loss. Loss of vision (grade 4) in the first patient occurred on Day 1 of Cycle 7. Following treatment on Day 8 of Cycle 7, the patient discontinued study-drug administration. Loss of vision in this patient was acute in onset, progressive, and had not resolved at the time of last follow-up. Significantly decreased light sensitivity throughout both eyes was reported with severe color loss. The patient was evaluated by a neuro-ophthalmologist and dense bilateral central scotomas were present. Assessment of visual acuity found the patient to be legally blind, although demonstrating excellent response to confrontation in the peripheral visual fields. Over a 2-month period the vision continued to deteriorate to nearly complete blindness. Retinal cone dysfunction was considered a less likely etiology for the vision loss, as a full-field electroretinogram was normal when the patient was almost totally blind, and extensive pallor and cupping had developed. In view of these findings, optic-nerve dysfunction was considered a more likely etiology for the vision loss.
A second patient experienced visual disturbances on Day 1 of treatment Cycle 1. A similar pattern of optic-nerve abnormality was observed with central scotomas. This patient had no progression of the visual symptoms and experienced partial recovery. The patient discontinued the study by Day 52. Both instances of vision loss were considered as possibly or probably related to E7107.
Pharmacokinetics
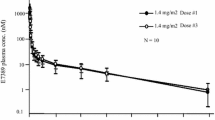
The PK parameters and mean plasma concentration-time curves for E7107 on Days 1 and 8 of Cycle 1 are presented in Table 4 and Fig. 1, respectively. Exposure to E7107, as assessed by C max and AUC, exhibited inter-patient variation but was similar on Days 1 and 8, and increased in a dose-related manner. The dose-normalized model-based geometric mean ratio for E7107 was contained completely within the 90 % confidence interval (0.68, 1.47) for C max, AUC0-t , and AUC0-∞, indicating dose proportionality on Day 1 and Day 8 for the dose range 0.6–5.7 mg/m2 used in this study. E7107 was rapidly distributed in plasma on both Day 1 and Day 8 (t max 0.32–0.44 h from the start of infusion on Day 1 and 0.33–0.42 h on Day 8; Fig. 1), demonstrated a moderate elimination t ½ and high CL, which did not change with increasing dose but was variable between doses. E7107 had a large V ss, which tended to decrease with increasing dose. On average, <10 % of E7107 was excreted in urine across all dose groups. Systemic exposure of the M8 metabolite of E7107 was negligible.

Mean plasma concentration of E7107 versus time in patients treated with a 30-min infusion on a Day 1 and b Day 8
Pharmacodynamics
Modulation of gene splicing in PBMCs was investigated as a potential biomarker for target engagement. The number of unspliced introns demonstrating a twofold change in expression level from baseline demonstrated saturation as the dose of E7107 increased. This was observed at both 6 and 24 h post-infusion; however, the number of unspliced introns decreased from 6 to 24 h (Fig. 2). Similar observations were made for unspliced introns, which demonstrated 5- and 10-fold changes in expression from baseline.

Unspliced introns with two-fold expression changes from baseline versus E7107 dose at a 6 h and b 24 h post-infusion
Efficacy
All 26 patients treated were included in the evaluation of response. Tumor response by RECIST was available for the 22 patients who remained on treatment at first restaging (end of Cycle 2). Of the four remaining patients, one withdrew because of AEs, one was lost to follow-up, one was taken off treatment by the physician because of clinical deterioration, and one experienced clinical progression prior to restaging. The best overall responses were: SD, eight patients (31 %), four (15 %) of whom had SD ≥4 months (adrenal cortical, papillary thyroid, pancreatic, non-small cell carcinoma). One patient with pancreatic cancer had a best response of 10 % decrease in the sum of longest tumor diameter from baseline and a reduction in cancer antigen 19–9 (CA19-9) levels, and remained on E7107 treatment for seven cycles until the patient developed vision loss.
Discussion
E7107 administered by intravenous infusion on Days 1 and 8 was well tolerated up to doses of 4.3 mg/m2 other than the visual disturbances described. The primary DLTs were MI and diarrhea. The most common AEs noted were nausea (76.9 %), vomiting (38.5 %), diarrhea (34.6 %), and fatigue (30.8 %).
A unique AE noted was vision loss, experienced by one patient each at E7107 3.2 mg/m2 and 4.3 mg/m2. The exact underlying mechanism for the visual toxicity remains unclear; however, in the patient with permanent visual loss, it is believed that the optic nerve was the target of the toxicity. The ability of E7107 to inhibit spliceosomes may be a possible explanation of the vision loss seen in some of these patients. In PBMCs from patients in this study, an increase in unspliced introns was clearly shown and suggests that E7107 may globally inhibit spliceosomes. Although the effects of this inhibition on the optic nerve are not known, there is some evidence in the literature to support the role of splicing factors in the retinal disorder retinitis pigmentosa. Several known genes that encode for spliceosomes are known to be mutated and involved in retinitis pigmentosa development, such as PRPF3, PRPF8, PRPF31, and Pim-1 associated proteins (PAP-1) [12–14]. Mutations in these splicing genes, such as PRPF31, can lead to dysfunctional and insoluble PRPF31 protein aggregates, leading to apoptosis in photoreceptor cells [15]. The ability of E7107 to inhibit spliceosomes may have had a functional effect similar to retinitis pigmentosa. The patient treated with E7107 3.2 mg/m2 exhibited trace retinal pigment epithelial changes and scotoma suggestive of cone dysfunction. The patient treated with E7107 4.3 mg/m2 had no evidence of retinitis and did not require retinal function tests.
Based on mean plasma concentrations, E7107 was rapidly distributed and was eliminated with a moderate mean t ½ (from 5.8 to 13.3 h) on Day 1 and Day 8. Mean C max and AUC showed some variation between patients, but were generally comparable on Day 1 and Day 8 for each patient. Mean C max and AUC for E7107 was dose-proportional on Day 1 and Day 8. Mean CL was high and generally remained unchanged with increasing dose, but variability was observed between doses. V ss was large and tended to decrease with increasing dose. The F e0–48 showed less than 10 % excreted in urine for all dose groups and generally remained unchanged with increasing dose, but exhibited large variability.
Efficacy was limited and did not reflect the extraordinary activity that was noted in the preclinical models. One reason may have been that the study was not designed to select patients who may have been most amenable to the antitumor mechanism of E7107. A full understanding of the underlying mechanisms of the compound needs to be elucidated; however, flow cytometry of cells treated with E7107 demonstrated cell-cycle arrest at both the G1 and G2/M phases, and that it is most effective against tumors with a functional loss of Rb increased p16 and cyclin E expression [4]. Moreover, increasing data suggest that somatically acquired mutations in the SF3B1 gene, which encodes a central component of the RNA slicing machinery, may be involved in several malignancies, specifically myelodysplastic syndromes, chronic lymphocytic leukemia, and refractory anemia with ringed sideroblasts [16–20]. E7107 has been shown to target SF3B1, suggesting that subsets of tumors with SF3B1 mutations may demonstrate efficacy [1]. Further studies are needed to determine whether SF3B1 mutations are present in solid tumors and their potential clinical significance.
In conclusion, E7107 is a novel first-in-class molecule that acts to inhibit spliceosome assembly and subsequent maturation of pre-mRNA. The drug was generally well tolerated with manageable systemic side effects; however, vision loss was noted in a small number of patients and led to study termination. Preclinical studies are ongoing to identify potential mechanisms for the observed vision loss.
References
Folco EG, Coil KE, Reed R (2011) The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25:440–444. doi:10.1101/gad.2009411
Kotake Y, Sagane K, Owa T (2007) Splicing factor SF3b as the molecular target of novel antitumor agent pladienolide [abstract 8211]. In: Program and abstracts of the Twelfth Annual Meeting of the RNA Society, Madison, WI, May 29–June 3
van Alphen RJ, Wiemer EA, Burger H, Eskens FA (2009) The spliceosome as target for anticancer treatment. Br J Cancer 100:228–232. doi:10.1038/sj.bjc.6604801
Uenaka T, Iwata M, Ozawa Y, Shimizu H, Kotake Y, Mizui Y, Yoshimatsu K, Asada M (2004) E7107, a new 7-urethane derivative of pladienolide D: correlation of the profile of cell cycle regulatory molecules with tumor cells’ response [abstract 2991]. Proc Am Assoc Cancer Res 45:691
Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 57:188–196. doi:10.7164/antibiotics.57.188
Iwata M, Ozawa Y, Uenaka T, Shimizu H, Niijima J, Kanada RM, Fukuda Y, Nagai M, Kotake Y, Yoshida M, Tsuchida T, Mizui Y, Yoshimatsu K, Asada M (2004) E7107, a new 7-urethane derivative of pladienolide D, displays curative effect against several human tumor xenografts [abstract 2989]. Proc Am Assoc Cancer Res 45:691
Niijima J, Kotake Y, Kanada RM, Nagai M, Fukuda Y, Sakai T, Ishihara H, Yoshida M, Tsuchida T, Iwata M, Uenaka T, Mizui Y, Abe S, Yoshimatsu K, Asada M (2004) E7107, a new 7-urethane derivative of pladienolide D: Discovery of a novel antitumor agent [abstract 2988]. Proc Am Assoc Cancer Res 45:691
Simon R, Freidlin B, Rubinstein L, Arbuck SG, Collins J, Christian MC (1997) Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 89:1138–1147. doi:10.1093/jnci/89.15.1138
Eisenhauer EA, O’Dwyer PJ, Christian M, Humphrey JS (2000) Phase I clinical trial design in cancer drug development. J Clin Oncol 18:684–692
Affymetrix Inc. (2005) Technical note: GeneChip® exon array design. http://media.affymetrix.com/support/technical/technotes/exon_array_design_technote.pdf. Accessed 25 July 2013
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216. doi:10.1093/jnci/92.3.205
McKie AB, McHale JC, Keen TJ, Tarttelin EE, Goliath R, van Lith-Verhoeven JJ, Greenberg J, Ramesar RS, Hoyng CB, Cremers FP, Mackey DA, Bhattacharya SS, Bird AC, Markham AF, Inglehearn CF (2001) Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet 10:1555–1562. doi:10.1093/hmg/10.15.1555
Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS (2001) A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell 8:375–381. doi:10.1016/S1097-2765(01)00305-7
Chakarova CF, Hims MM, Bolz H, Abu-Safieh L, Patel RJ, Papaioannou MG, Inglehearn CF, Keen TJ, Willis C, Moore AT, Rosenberg T, Webster AR, Bird AC, Gal A, Hunt D, Vithana EN, Bhattacharya SS (2002) Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet 11:87–92. doi:10.1093/hmg/11.1.87
Deery EC, Vithana EN, Newbold RJ, Gallon VA, Bhattacharya SS, Warren MJ, Hunt DM, Wilkie SE (2002) Disease mechanism for retinitis pigmentosa (RP11) caused by mutations in the splicing factor gene PRPF31. Hum Mol Genet 11:3209–3219. doi:10.1093/hmg/11.25.3209
Rossi D, Bruscaggin A, Spina V, Rasi S, Khiabanian H, Messina M, Fangazio M, Vaisitti T, Monti S, Chiaretti S, Guarini A, Del Giudice I, Cerri M, Cresta S, Deambrogi C, Gargiulo E, Gattei V, Forconi F, Bertoni F, Deaglio S, Rabadan R, Pasqualucci L, Foá R, Dalla-Favera R, Gaidano G (2011) Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood 118:6904–6908. doi:10.1182/blood-2011-08-373159
Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, Steensma DP, Pardanani A, Hanson CA, Tefferi A (2012) SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 119:569–572. doi:10.1182/blood-2011-09-377994
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ, Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365:1384–1395. doi:10.1056/NEJMoa1103283
Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, O’Keefe C, Rogers HJ, Sekeres MA, Maciejewski JP, Tiu RV (2012) SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26:542–545. doi:10.1038/leu.2011.232
Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F, Stamatoullas-Bastard A, Tanguy-Schmidt A, Beyne-Rauzy O, de Botton S, Guerci-Bresler A, Göhring G, Schlegelberger B, Ganser A, Bernard OA, Fontenay M, Heuser M (2012) SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia 26:1137–1140. doi:10.1038/leu.2011.321
Acknowledgments
We gratefully acknowledge the participating patients, their families, and study investigators for their invaluable contribution. Editorial support was provided by Deborah McGregor, PhD, of Complete Medical Communications, funded by Eisai Inc., and Jo Ann of the Department of Investigational Cancer Therapeutics, University of Texas M.D. Anderson Cancer Center, Houston, Texas. This study was funded by Eisai Inc.
Authors’ contributions
All authors were involved in the writing, review, and/or revision of the manuscript, and have approved the manuscript for submission. D. Hong and P. LoRusso were principal investigators for the study and were involved in the development of the methodology, the acquisition of data, and the analysis and interpretation of the results. R. Kurzrock, A. Naing, J. Wheler, G. Falchook, J. Schiffman, N. Faulkner, and M. J. Pilat were involved in the acquisition of data. J. O’Brien was involved in the development of the methodology, and the analysis and interpretation of the results.
Financial support
This study was funded by Eisai Inc.
Conflicts of interest
D. S. Hong received research grant funding from Eisai for this study. J. O’Brien is affiliated with Eisai Inc., NJ, USA. The remaining authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
The submission is original work not previously published in any substantial part, and is not under consideration for publication elsewhere.
The manuscript has been read and approved for submission by all authors.
Rights and permissions
About this article
Cite this article
Hong, D.S., Kurzrock, R., Naing, A. et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 32, 436–444 (2014). https://doi.org/10.1007/s10637-013-0046-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-0046-5