
Summary
Aim This phase I/II study of saracatinib in combination with gemcitabine in patients with advanced pancreatic cancer was conducted by the NCIC Clinical Trials Group. The aims were to define the recommended phase II dose (RP2D) of saracatinib when combined with gemcitabine, and assess the efficacy of this combination in advanced pancreatic cancer. Patients and Methods Eligibility criteria included locally advanced or metastatic pancreatic adenocarcinoma and no prior chemotherapy. In phase I saracatinib was escalated in combination with gemcitabine (1000 mg/m2) to determine the recommended phase II dose (RP2D). The study was then expanded to a single arm phase II trial using a Simon 2-stage design. The primary endpoint was objective tumor response (OR) plus stable disease ≥4 months (SD4) rate; if ≥8 patients had OR+SD4, the study would proceed to stage 2. Results Thirteen patients were enrolled into the phase I portion of this study. Saracatinib 175 mg PO daily was chosen as the RP2D in combination with gemcitabine. Twenty-one additional patients were then enrolled at the RP2D (phase II). Of the 22 response evaluable patients treated at the RP2D, 9 patients (40.9%) had progressive disease, 6 patients (27.3%) had stable disease for less than 4 months, 5 patients (22.7%) had SD4, and 2 patients (9.1%) had a partial response to treatment. Objective criteria for continuing to stage 2 were thus not met and the trial was closed following the accrual of 34 patients. Conclusion Saracatinib 175 mg daily in combination with gemcitabine is well tolerated but the combination did not improve efficacy over what would be expected from gemcitabine alone.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer is the fourth leading cause of cancer death in North America [1]. Median survival in advanced disease is approximately 6 months [2], response rates to cytotoxic chemotherapy are low, and 1-year survival with treatment is less than 25% [3]. Gemcitabine has been regarded as the standard backbone of systemic therapy for pancreatic cancer based upon a 1997 trial comparing gemcitabine versus fluorouracil that demonstrated an improvement in median and one-year survival [4].
Numerous trials undertaken using cytotoxic agents in combination with gemcitabine have not improved overall survival [5]. Tyrosine kinases involved in tumorigenesis represent promising targets, and inhibition of the epidermal growth factor receptor tyrosine kinase in combination with gemcitabine was shown to modestly improve survival compared with gemcitabine alone [3].
Src is a non-receptor tyrosine kinase protein product of the proto-oncogene c-Src, the cellular homolog of the Rous sarcoma virus transforming protein v-Src [6]. Src is involved in the formation of focal adhesions and cellular migration, and is thought to have an important role in tumorigenesis, invasion, and metastasis [7]. Src has been shown to be overexpressed in over 70% of pancreatic carcinoma cell lines [8]. Saracatinib (AZD0530), (N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl) ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine), (AstraZeneca) is an orally bioavailable aniline-quinazoline that functions as an inhibitor of the Src tyrosine kinase. In experimental models it has been shown to inhibit the phosphorylation of paxillin and focal adhesion kinase, which are thought to be critical in modulating cell motility and invasion [9]. A phase I study conducted with saracatinib revealed that dose limiting toxicities (DLTs) occurred at doses above 175 mg PO daily (leukopenia, septic shock with renal failure, asthenia, febrile neutropenia, and dyspnea)[10]. Preclinical studies have shown that human pancreatic adenocarcinoma cells with constitutively active Src have gemcitabine chemoresistance [11]. In nude mice orthotopic xenografts, Src inhibition was found to enhance gemcitabine cytotoxicity, and significantly decrease tumor growth and inhibit metastases [11].
The NCIC clinical trials group undertook a phase I/II study of saracatinib in combination with gemcitabine as first line therapy for patients with advanced pancreatic cancer to evaluate the safety, tolerability, toxicity profile, and efficacy of this combination.
Patients and methods
Patient selection
The study population consisted of patients with unresectable, locally advanced or metastatic pancreatic cancer who had not received prior chemotherapy, other than 5-fluorouracil (± leucovorin) or gemcitabine given concurrently with radiation treatment as a radiosensitizer. The institutional review boards of the participating institutions approved the study, and all patients provided written informed consent.
Patients were required to be ≥18 years of age, Eastern Cooperative Oncology Group (ECOG) performance status 0, 1 or 2; absolute granulocyte count ≥1.5 × 109/L, platelet count ≥100 × 109/L, and normal serum creatinine and bilirubin. Aspartate aminotransferase (AST) and alanine transaminase (ALT) were required to be ≤2.0 × the upper limit of normal (ULN), unless liver metastases were present (≤5 × ULN). Patients enrolled to the phase II portion of the study were required to have measurable disease (by Response Evaluation Criteria in Solid Tumors [RECIST]).
Exclusion criteria included concurrent other malignancies, any serious underlying medical conditions that would impair the ability of the patient to receive protocol treatment, or a gastrointestinal tract disease resulting in an ability to take oral medication.
Study design
There was an initial dose-finding phase I study of saracatinib, in combination with the standard dose and schedule of gemcitabine. The study was then expanded at the recommended phase two dose (RP2D) of saracatinib in combination with gemcitabine to a single arm phase II study in a two-stage Simon design. AstraZeneca provided the study drug. NCT Registration ID: NCT00265876.
Phase I
The starting dose of saracatinib was 100 mg PO daily. Gemcitabine was given at 1000 mg/m2 over 30 min weekly for 3 weeks followed by one-week break. Doses of saracatinib were escalated to determine the RPTD when given in combination with full dose gemcitabine. A minimum of 3 patients were to be entered on each dose level. Dose limiting toxicities (DLTs) were defined as follows: creatinine ≥ Grade 2, proteinuria or hematuria ≥ Grade 3, Hypotension ≥ Grade 3, other Grade 3 or 4 adverse events not clearly attributable to gemcitabine, other toxicities of concern to the investigator or NCIC CTG, or the inability to administer saracatinib for more than 14 days due to toxicity.
If 0/3 patients exhibited a DLT the dose level was increased. If 1/3 patients exhibited DLT at a given dose level, this cohort would be expanded to 6 evaluable patients. If 2/3 or 2/6 patients exhibit DLT at a given dose level, this dose would be considered the maximum administered dose (MAD) and the RP2D would be the previous dose level.
Phase II
Once the RP2D level of saracatinib was defined, this was given daily with gemcitabine at standard dose (1000 mg/m2 every week for 3 weeks followed by 1 week break).
Patient evaluation
Baseline radiological investigations to document disease were done within 21 days prior to study treatment. The study was amended to include pulmonary function tests (PFTs) and high resolution CT scan of the chest at baseline and at the end of cycle 1, due to concerns about pulmonary adverse events possibly associated with saracatinib noted in a previous study (12); the study required PFTs to be ≥80% of predicted for patients enrolled up until June 2007. Radiological assessments for tumor measurements were conducted after every second cycle (every 8 weeks).
Duration of therapy
Study treatment continued until unacceptable toxicity, patient request, or progression. Gemcitabine was only to be continued until 2 cycles beyond complete or stable partial response criteria were met, and for a maximum of 6 cycles in case of stable disease, following which saracatinib alone continued until unacceptable toxicity or disease progression.
Dose modifications for saracatinib
For grade 2 toxicity not immediately resolving with symptomatic treatment, saracatinib was held until the toxicity improved to ≤ grade 1 and then resumed without dose reduction. On second occurrence, the dose was reduced by 25%. For grade 3 toxicity, saracatinib was withheld until ≤ grade 1 and then resumed at a 25% dose reduction. For grade 4 toxicity protocol, therapy was discontinued.
Pharmacokinetics
In the phase I portion of the study, gemcitabine pharmacokinetics were assessed on day 15 of cycle 1 at the following time points: pre-infusion, 15 min after start of infusion, just prior to the end of infusion, and 5, 10, 15, and 30 min, and 1, 2, 4, 6, and 24 h post infusion. Blood samples for saracatinib were collected on day 15 of cycle 1, pre-dose, and 2, 4, 6, 8 and 24 h post dosing. In the phase II portion, in addition to the day 15 collections, blood samples were also collected at day 1 of cycle 1 for gemcitabine and day 14 of cycle 1 for saracatinib at same time points listed above.
Plasma concentrations of gemcitabine and saracatinib were quantitated with validated HPLC-tandem mass spectrometry methods. Pharmacokinetic parameters were calculated by non-compartmental methods using the WinNonlin Version 5.1 (Pharsight Corp., Mountain View, CA). Pharmacokinetic variables were analyzed with descriptive statistics. Participation in the pharmacokinetic portion of the study was mandatory for the phase I component of the study, and optional for the phase II component.
Statistical methods
The primary endpoint of the phase I portion of the study was to determine the RP2D dose of daily oral saracatinib when given in combination with standard gemcitabine chemotherapy. It was planned that patients treated at the RP2D in the phase I portion would be pooled with the phase II patients in the efficacy analyses. In the phase II study the primary endpoint was to determine objective tumor response plus prolonged stable disease rate (defined as partial or complete response as defined by the RECIST criteria or confirmed stable disease of 4 months) (OR/SD4). Secondary endpoints included overall survival, response or stable disease duration rate, progression-free survival, and toxicity.
The optimal Simon two-stage phase II design was used with results indicating lack of efficacy resulting in study termination after stage 1 [12]. The treatment combination was assumed to be inactive if the objective response plus prolonged stable disease rate was at most 30% and active if it was at least 50%. This was based on an objective response/stable disease rate of over 50% observed for the gemcitabine arm in a previous study [13]. The first stage involved the accrual of 22 response evaluable patients and if at least 8 of these patients had an OR/SD4, the study would proceed to stage II. To qualify as being response evaluable patients must have received at least one cycle of therapy and have their disease re-evaluated (with the exception of patients that exhibit objective clinical progression prior to the end of cycle 1, who will also be considered evaluable). The second stage involved accrual of an additional 24 response evaluable patients. If 18 or more of the 46 total response evaluable patients had an OR/SD4, the treatment would be deemed active. The cut-off points were calculated taking into account the alpha and power for this design. The alpha using this design is 0.097, with a power of 0.91.
The use of a multinomial stopping rule in a single arm phase II study has previously been validated [14, 15]. This approach allows for assessment of tumor response and early progression, and thus may represent a more efficient stopping rule compared with monitoring tumor response alone.
An exploratory analysis was performed to examine changes in tumor size as a continuous variable [16, 17]. The logarithm of the sum of the longest diameters of target lesions was calculated at baseline and at 8 weeks. The change in tumor size from this study was compared to the change in tumor size for the gemcitabine only arm of the PA.1 [13] and PA.3 (3) studies using the Wilcoxon rank sum test.
Results
Phase I
Thirteen patients were enrolled into the phase 1 portion (Table 1). The median number of cycles administered was 2. Nine phase I patients went off study because of objective progression, two patients due to toxicity (grade 2 vital capacity changes, grade 2 pneumonitis), one patient refused further treatment, and one patient was eventually removed from protocol treatment due to ineligibility (diagnosis corrected to neuroendocrine tumor 9 months after study entry). Three patients each were accrued at the first dose level of saracatinib 100 mg PO daily and the second level of 150 mg PO daily with no DLT. Seven patients were then accrued at saracatinib 175 mg PO daily. No patient experienced a DLT at this dose level. However as saracatinib doses above 175 mg were found to be intolerable in the phase I single agent study (leucopenia, asthenia, septic shock with renal failure at 250 mg; febrile neutropenia, dyspnea at 200 mg) [10], the decision was made to declare 175 mg PO daily the RP2D for saracatinib in combination with gemcitabine. Adverse events are summarized in Table 2.
All patients enrolled in the phase I portion of the study had pulmonary function testing performed at baseline and at the end of cycle 1. There were no grade 3 or 4 toxicities seen for carbon monoxide diffusing capacity (DLCO), forced expiratory volume (FEV) or forced vital capacity (FVC). Forty-six percent (6/13) of patients had a decrease in DLCO by one grade (four patients from grade 0 to grade 1, two patients from grade 1 to grade 2). Eight percent (1/13) of patients had a decrease in FEV by one grade (one patient from grade 0 to grade 1). Eight percent (1/13) of patients had a decrease in FVC by one grade (one patient from grade 1 to grade 2).
Phase II
Twenty-one patients were enrolled in the phase II portion of this study. When combined with the 7 patients enrolled at the RP2D in the phase I portion, there were 28 patients enrolled and treated at the RP2D. Of the 28 patients treated at the RP2D, 16 patients had metastatic disease, 11 patients had locally advanced disease, and 1 patient was found to be ineligible due to neuroendocrine pathology. The majority (22/28) were performance status 1. For the 28 patients treated at the RP2D, the median number of cycles administered was 3 (range 1–18). One patient required a saracatinib dose reduction due to hematologic toxicity and 12 patients required a gemcitabine dose reduction due to toxicity (hematologic, biochemical, and skin toxicity). Thirteen (of 28) patients went off study because of objective progression, 3 patients died of disease while on study, 4 patients refused further treatment, 4 patients went off treatment as a result of adverse events considered possibly related to saracatinib and/or gemcitabine study treatment (two pneumonitis (grade 2 and grade 3), one grade 2 nausea, one grade 2 reduction in forced vital capacity), 2 patients went off due to symptomatic progression, 1 patient ended protocol treatment because of an intercurrent illness, and 1 patient was removed due to ineligibility (neuroendocrine pathology).
Safety
Grade 3 and 4 non-hematologic adverse events considered possibly related to saracatinib and/or gemcitabine observed in the 28 patients treated at the RP2D level were: fatigue (14%), anorexia (7%), diarrhea (4%), gastrointestinal hemorrhage (4%) and pneumonitis (4%) (Table 2). Six patients (21%) had grade 3 or 4 granulocytopenia, 6 patients (21%) had grade 3 or 4 anemia, and 5 patients (18%) experienced grade 3 thrombocytopenia (Table 2). Two patients (7%) had a grade 3 elevation in AST and 4 patients (14%) had a grade 3 elevation in bilirubin. Among the 28 patients treated at the RP2D, there were 3 serious adverse events reported which were assessed to be related to protocol treatment. One patient was hospitalized after evaluations at the end of cycle 3 revealed progressive disease, a left ventricular ejection fraction of 48% (considered possibly related to saracatinib and gemcitabine) and thrombus in the right atrium. Another patient with fever, cough and dyspnea on at the end of cycle 2 was hospitalized and removed from the study after imaging demonstrated bilateral interstitial pneumonitis (considered possibly related to both saracatinib and gemcitabine) and pulmonary emboli. A third patient was hospitalized for gastrointestinal bleed with grade 3 thrombocytopenia thought to be possibly related to saracatinib.
Of the 28 patients enrolled at the RP2D, pulmonary function testing was performed at baseline and at the end of cycle 1 in 13 patients, and data were available for 12 patients. There were no grade 3 or grade 4 toxicities seen for DLCO, FEV or FVC. Eight percent (1/12) of patients had a decrease in DLCO by one grade (one patient from grade 0 to grade 1). Seventeen percent (2/12) of patients had a decrease in FEV by one grade (one patient from grade 0 to grade 1, one patient from grade 1 to grade 2). Seventeen percent (2/12) of patients had a decrease in FVC by one grade (one patient from grade 0 to grade 1, one patient from grade 1 to grade 2).
Efficacy
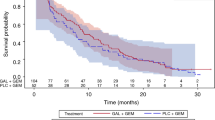
Of the 27 eligible patients treated at the RP2D of saracatinib 175 mg PO daily (1 patient ineligible due to neuroendocrine pathology), 22 patients were evaluable for response (reasons inevaluable: 3 patients received less than 1 cycle of treatment, 2 patients did not have disease reassessed as per protocol), 9 patients (40.9%) had progressive disease, 6 patients (27.3%) had stable disease for less than 4 months, 5 patients (22.7%) had stable disease for 4 months or longer (median duration 7.4 months, range 5.5–16.6 months), and 2 patients (9.1%) had a partial response to treatment (duration 3.6 and 7.6 months). The response plus stable disease for 4 months (OR/SD4) rate was 7/27 (26%; (95% CI: 11.1,46.3%)). The predefined criteria for continuing phase II accrual (at least 8 patients with OR/SD4) were not met and therefore the trial was closed. Of the 27 eligible patients that were treated at the saracatinib dose of 175 mg per day, the median overall survival was 6.2 months (95% CI, 3.68–7.79) (Fig. 1).

Kaplan Meier curve of overall survival for patients treated at the saracatinib dose of 175 mg per day
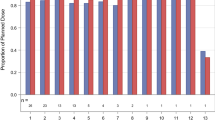
Exploratory analyses examining individual changes in tumor size were also performed. In this study, the mean for the changes in the logarithm of total tumor size from baseline was −0.031 (standard deviation = 0.16). This change was not significantly different when compared to mean tumor size changes in the gemcitabine alone arm of the NCIC CTG PA.1 phase 3 study of gemcitabine versus the MMP inhibitor BAY-12-9566, [13] (mean change = −0.066, p = 0.47), but is significantly less then what was observed from the gemcitabine alone arm in the NCIC CTG PA.3 phase 3 study of gemcitabine with or without erlotinib (3) (mean change −0.114, p = 0.02). Waterfall plots were also drawn, comparing results from this study with the PA.3 study (Fig. 2).

Waterfall plot of changes in tumor size for the combination of saracatinib and gemcitabine in this study (black) and the gemcitabine alone from the PA.3 study (red)
Pharmacokinetics
Pharmacokinetics of gemcitabine
Pharmacokinetic data for gemcitabine was available from 28 patients. The day 15 mean area under the curve (AUC) for gemcitabine was 6483 ± 1538 ng/ml (n = 3), 7275 ± 594 ng/ml (n = 3), and 8096 ± 4346 ng/ml (n = 14) at saracatinib doses of 100 mg, 150 mg, and 175 mg respectively. These values were not significantly different, and are similar to levels reported in the literature [18]. The mean AUC of gemcitabine alone was 7775 ± 3079 ng/ml (n = 15) compared with 8096 ± 4346 ng/ml (n = 14) for gemcitabine in the presence of saracatinib, which were not significantly different.
Pharmacokinetics of saracatinib
Pharmacokinetic data for saracatinib was available for 11 patients. The day 15 mean AUC for saracatinib (in the presence of gemcitabine) was 1877 ± 0 ng/ml (n = 1), 4267 ± 2668 ng/ml (n = 2), and 8967 ± 4875 ng/ml (n = 8) at saracatinib doses of 100 mg, 150 mg, and 175 mg respectively. The mean AUC of saracatinib alone at the 175 mg dose was 4449 ± 2574 ng/ml (n = 3), which was not significantly different to the mean AUC of saracatinib in the presence of gemcitabine 8967 ± 4875 ng/ml, although this comparison is limited due to the small number of samples of saracatinib alone.
Discussion
Successful drug development for advanced pancreatic cancer continues to represent a challenge. Src is a tyrosine kinase that has been implicated in tumorigenesis, invasion, and metastasis [7], and pre-clinical data suggests that Src inhibition may have anti-tumor activity in pancreatic cancer. In addition there is evidence to suggest that inhibition of Src may work synergistically with gemcitabine, potentially through saracatinib mediated inhibition of ribonucleotide reductase activity (an enzyme that when overexpressed may contribute to gemcitabine resistance) [11].
Saracatinib and gemcitabine can be administered together safely using the maximally tolerated doses of both drugs, and this combination was well tolerated at the RP2D with few grade 3 adverse events and only 4 (14.3%) grade 4 adverse events (granulocytopenia and anemia). Of note, although there were no grade 3 or 4 changes in pulmonary function, a significant number of patients did have slight worsening in pulmonary function testing after one month of treatment. As there was no gemcitabine alone arm, it is unclear to what extent saracatinib contributed to these findings. Nonetheless, it will be important to closely monitor pulmonary function in future trials with this agent.
The primary purpose of a phase II study is to define agents that warrant further study and screen out those that are ineffective. As the response rate to gemcitabine in pancreatic cancer is low, and the survival benefit is associated with a delay in progression we selected a combination of objective response and sustained (≥4 months) stable disease (OR/SD4) as the primary outcome of interest. We selected a single arm design, using our own historical data from the NCIC CTG PA.1 [13] and NCIC CTG PA.3 [3] studies to define outcomes of interest. After the first stage of enrolment, 5/27 of the response evaluable patients had stable disease for over 4 months, and 2/27 having a partial response, thus resulting in an OR/SR4 of 26% (7/27; 95% CI: 11.1, 46.3%). These efficacy results did not meet the predefined criteria for continuing accrual to this study. The overall rate of disease control (including partial response and stable disease (of any duration)) was 13/27 (48%), which is comparable to that seen in the PA.1 gemcitabine arm (59%) [13], and the PA.3 gemcitabine arm (49%) [3]. Of note, while the entry criteria for all 3 studies were very similar, the inclusion criteria for the present study were more stringent due to the requirement for normal pulmonary function testing (which was not required in the previous studies).
Exploratory analyses were performed to examine changes in tumor size as a continuous variable. The categorical characterization of tumor response using RECIST criteria can lead to overlooking potentially important results (i.e. responses that do not meet partial response criteria), which are captured when change in tumor size is assessed as a continuous variable [17]. This type of analysis has been suggested to be a potentially more sensitive early determinant of treatment effect (or lack of effect) [17]. A comparison of the changes in tumor size observed in this study with the changes in tumor size in the gemcitabine alone arm of the PA.1 study revealed no significant differences. In comparison to the gemcitabine alone arm of the PA.3 study, there appeared to be less changes in tumor size in this study. This may be due to differences in the patient populations between PA.3 and our study (a smaller proportion of patients with locally advanced disease were present in the PA.3 study compared with this study). These exploratory analyses support our conclusion that the addition of saracatinib to gemcitabine does not appear to improve response compared to what would be expected from gemcitabine alone.
The pharmacokinetic analysis revealed that the exposure of gemcitabine does not appear to be effected by the presence of saracatinib. The exposure to saracatinib was found to increase with dose, but with large inter-subject variability.
It remains unclear why targeted agents with activity in pancreatic cancer pre-clinical models, have not improved outcomes in patients. Better pre-clinical models are needed and this work is ongoing [19]. It is also likely there is a greater level of complexity to the molecular pathways involved in tumorigenesis and metastasis than presently understood. This is particularly relevant in pancreatic cancer where a high level of genetic mutations clustered around 12 key signaling pathways has been seen [20]. In addition, potentially agents have benefit in certain molecularly defined subsets of pancreatic cancer, and appropriate biomarkers could be used to select for a subgroup of patients that may be responsive [21]. Finally, given that Src is thought to be involved in cellular migration, invasion and metastasis, it is possible that benefit from Src inhibitors may be more profound in the adjuvant or locally advanced settings, in an effort to prevent the development of metastasis, rather than in the setting of already established metastatic disease.
In summary, the combination of saracatinib and gemcitabine was well tolerated at the RP2D dose of saracatinib 175 mg PO daily in combination with standard doses of gemcitabine, but did not appear to improve efficacy over what would be expected from gemcitabine alone.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin. doi:10.3322/CA.2007.0010
Glimelius B, Hoffman K, Sjoden PO, Jacobsson G, Sellstrom H, Enander LK, Linne T, Svensson C (1996) Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann Oncol
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, National Cancer Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. doi:10.1200/JCO.2006.07.9525
Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol
Yip D, Karapetis C, Strickland A, Steer CB, Goldstein D (2006) Chemotherapy and radiotherapy for inoperable advanced pancreatic cancer. Cochrane Database Syst Rev. doi:10.1002/14651858.CD002093.pub2
Martin GS (2001) The hunting of the Src. Nat Rev Mol Cell Biol. doi:10.1038/35073094
Boyer B, Bourgeois Y, Poupon MF (2002) Src kinase contributes to the metastatic spread of carcinoma cells. Oncogene. doi:10.1038/sj.onc.1205298
Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Luhrs H, Friess H, Buchler MW, Adler G (1998) Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. doi:10.1006/bbrc.1997.8043
Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-van der Brempt C, Morgentin R, Norman RA, Olivier A, Otterbein L, Ple PA, Warin N, Costello G (2006) N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. doi:10.1021/jm060434q
Tabernero J, Cervantes A, Hoekman K et al (2007) Phase I study of AZD0530, an oral potent inhibitor of Src kinase: First demonstration of inhibition of Src activity in human cancers. J Clin Oncol 25:Abst 3520
Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE (2004) Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res
Simon R (1989) Optimal two-stage designs for phase II clinical trials. Controlled Clinical Trials 1–10
Moore MJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, Hagan K, Greenberg B, Colwell B, Zee B, Tu D, Ottaway J, Humphrey R, Seymour L, National Cancer Institute of Canada Clinical Trials Group (2003) Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. doi:10.1200/JCO.2003.02.098
Zee B, Melnychuk D, Dancey J, Eisenhauer E (1999) Multinomial phase II cancer trials incorporating response and early progression. J Biopharm Stat
Dent S, Zee B, Dancey J, Hanauske A, Wanders J, Eisenhauer E (2001) Application of a new multinomial phase II stopping rule using response and early progression. J Clin Oncol
Karrison TG, Maitland ML, Stadler WM, Ratain MJ (2007) Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst. doi:10.1093/jnci/djm158
Dhani N, Tu D, Sargent DJ, Seymour L, Moore MJ (2009) Alternate endpoints for screening phase II studies. Clin Cancer Res. doi:10.1158/1078-0432.CCR-08-2034
Price TJ, Lipton L, McGreivy J, McCoy S, Sun YN, Rosenthal MA (2008) Safety and pharmacokinetics of motesanib in combination with gemcitabine for the treatment of patients with solid tumours. Br J Cancer. doi:10.1038/sj.bjc.6604723
Jimeno A, Solomon A, Karikari C et al (2008) A prospective validation of a direct tumor xenograft model in pancreatic ductal adenocarcinoma (PDA). J Clin Oncol 26:Abstr 4500
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. doi:10.1126/science.1164368
Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M (2009) Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. doi:10.1158/1078-0432.CCR-08-3021
Funding
This study was supported by funding from the Canadian Cancer Society. AstraZeneca provided saracatinib and partial funding to support the study.
Conflicts of Interest
Lesley Seymour received research funding from Astra Zeneca for this trial and has shares with Astra Zeneca. Derek Jonker attended an advisory board for Astra Zeneca.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Renouf, D.J., Moore, M.J., Hedley, D. et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Invest New Drugs 30, 779–786 (2012). https://doi.org/10.1007/s10637-010-9611-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-010-9611-3