
Summary
Background To define maximum-tolerated dose (MTD), dose-limiting toxicities (DLTs), and preliminary efficacy of sorafenib plus capecitabine/cisplatin in advanced gastric cancer (AGC) patients. Methods Four dose-level combinations were tested in a standard 3 + 3 dose escalation design. Level 1: sorafenib 400 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 80 mg/m2. Level 2: sorafenib 800 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 80 mg/m2. Level 3: sorafenib 800 mg/d, capecitabine 2,000 mg/m2/d, cisplatin 80 mg/m2. Level 1A: sorafenib 800 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 60 mg/m2. Results There were 1 DLT at Level 2, and 2 DLTs at Level 3 (Level 3 was MTD). Since the relative dose intensity (RDI) of sorafenib and capecitabine could not be maintained at Level 2, Level 1A was newly investigated. As no DLT was observed and RDI remained above 80%, Level 1A is the recommended dose for the next clinical trial. Objective response rate was 62.5% (10 of 16 patients, 95% CI; 38.8–86.2%). Median progression-free survival and overall survival were 10.0 months (95% CI; 7.4–13.8) and 14.7 months (95% CI; 12.0–20.0), respectively. Conclusions Sorafenib 400 mg bid daily, capecitabine 800 mg/m2 bid (days 1–14), and cisplatin 60 mg/m2 (day 1) is recommended for further development in AGC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastric cancer is the second most common cause of cancer death worldwide [24]. Palliative chemotherapy in metastatic or unresectable advanced gastric cancer (AGC) patients improves quality of life and overall survival compared with best supportive care [11, 27]. Even with the introduction of newer chemotherapeutic agents in recent years, treatment results from globally executed, randomized, well-controlled phase III trials have not improved, with overall response rates of 25–50% and overall survival of 8–12 months [4, 16, 36]. There is an urgent need for additional therapies with better response rates and increased overall survival.
Molecularly targeted therapies have been introduced for the treatment of AGC and a number of clinical trials with monoclonal antibodies against epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), and human epidermal growth factor receptor 2 (HER2) have been tried. A randomized phase III clinical trial of trastuzumab (ToGA trial) in combination with chemotherapy showed survival benefit over chemotherapy alone in patients with HER2-overexpressing gastric cancer [22, 26, 31, 35]. VEGF expression is a negative prognostic factor for survival in patients with gastric cancer and inhibition of VEGF activity by an immunoneutralizing antibody showed activity in a gastric cancer xenograft model [14, 37]. A recent phase III study of bevacizumab combined with capecitabine (or 5-FU) and cisplatin in advanced gastric cancer (AVAGAST trial) has failed to meet the primary endpoint of overall survival but showed significant advantage in overall response rate (ORR; 29.5% vs 38.0%, p value 0.0037) and progression-free survival (PFS: 5.3 months vs 6.7 months, p value 0.0121) over chemotherapy alone [15]. In addition, in many tumors, signal transduction of cellular proliferation and survival through transmembrane receptors (EGFR, HER2, c-Met) results in the activation of RAS and downstream RAF/MEK/ERK pathways; mitogen-activated protein kinase (MAPK) is activated in some tumors with mutations in the RAF pathway [1, 6, 23, 30].
Sorafenib (BAY 43-9006; Nexavar®) is a potent inhibitor of multiple intracellular (RAF/MAPK) and cell surface (cKIT, FLT-3, VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR-ß) kinases. Sorafenib is known to inhibit tumor growth in several ways, including direct inhibition of MAPK-dependent tumor cell proliferation and angiogenesis inhibition via VEGFR-1, VEGFR-2 and PDGFR-ß. Sorafenib monotherapy has been evaluated in the treatment of advanced hepatocellular carcinoma and clear-cell renal cell carcinoma and is currently approved for those indications [9, 20]. In several phase I studies, sorafenib was combined with oxaliplatin, doxorubicin, gemcitabine, paclitaxel, and carboplatin, combinations which showed acceptable safety profiles and evidence of activity in patients with refractory solid cancers [10, 19, 28, 32].
Capecitabine plus cisplatin (XP regimen) has become a standard 1st-line chemotherapy for AGC based on a recent phase III study demonstrating the non-inferiority of XP to 5-fluorouracil and cisplatin (FP), as assessed by progression-free survival (PFS) [16, 17]. Although there is a suggestion that triplet combinations might be more active than doublet combinations with better survival [4, 36], the considerable toxicities associated with triplets might be exacerbated by the addition of sorafenib. Therefore, in the present phase I dose-finding study in AGC patients, sorafenib was combined with the doublet XP regimen.
Patients and methods
Patient eligibility
Patients 18–75 years of age with pathologically documented gastric or gastroesophageal junction adenocarcinoma were eligible for inclusion in this study. They were required to have metastatic or unresectable disease, either initially diagnosed or relapsed after surgery, with no history of chemotherapy or radiotherapy. Measurable or evaluable lesions according to Response Evaluation Criteria in Solid Tumours (RECIST) were required for enrollment. Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status ≤2 and adequate organ function defined as: Hemoglobin ≥8.0 g/dL, absolute neutrophil count (ANC) ≥ 1,500/mm3, platelet count ≥ 100,000/mm3, total bilirubin ≤ 2.0 mg/dL, alanine aminotransferase (ALT) and aspartate aminotranferase (AST) ≤ three times the upper limit of normal (ULN) or ≤ five times the ULN in the case of liver metastasis, prothrombin time (international normalized ratio) ≤1.5, partial thromboplastin time ≤ 1.5 × ULN, and serum creatinine ≤1.5 mg/dL.
Patients with a history of or concurrent malignancies other than gastric cancer (except for curatively treated non-melanoma skin cancer or in situ carcinoma of the cervix uteri), obvious peritoneal seeding with bowel obstruction, serious gastrointestinal bleeding, peripheral neuropathy, significant neurologic or psychiatric disorders, seizure disorder requiring medication, clinically active serious infection, or other serious illness were excluded. Patients were excluded if they had cardiac diseases such as congestive heart failure (> New York Heart Association class 2), active coronary artery disease within 6 months, arrhythmias requiring anti-arrhythmic therapy except beta blockers or digoxin, and uncontrolled hypertension. Pregnant or lactating women and women of child-bearing potential not employing adequate contraception were also excluded.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines and the study protocols and amendments were reviewed and approved by the institutional review board of the Asan Medical Center (Seoul, Korea). All patients provided written informed consents before study entry.
Study design and treatment
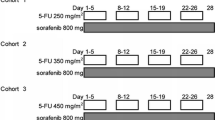
This was a single center, open-label, dose-escalation study to define the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs) and to assess the safety profile and preliminary clinical efficacy in patients with metastatic or unresectable AGC. Four dose levels of the sorafenib (p.o. bid, daily), capecitabine (p.o. bid on days 1–14), and cisplatin (i.v. on day 1) combination were tested.
-
Level 1
sorafenib 400 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 80 mg/m2
-
Level 2
sorafenib 800 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 80 mg/m2
-
Level 3
sorafenib 800 mg/d, capecitabine 2,000 mg/m2/d, cisplatin 80 mg/m2
-
Level 1A
sorafenib 800 mg/d, capecitabine 1,600 mg/m2/d, cisplatin 60 mg/m2.
Patients received up to 8 cycles of chemotherapy (3 weeks/cycle) and, on completing the planned chemotherapy, non-progressing patients were allowed to continue sorafenib monotherapy at the same dose and schedule until disease progression or intolerable toxicity occured.
A standard 3 + 3 dose escalation design was used. Three patients were treated in each dose level cohort. If one patient experienced DLT, three additional patients were added to that cohort. If none of the three patients or less than two of six patients in each cohort experienced DLT, the dose was escalated to the next level until the MTD was determined or the maximum dose level (Level 3) was reached in the absence of a DLT. The MTD was defined as the dose where two or more patients out of six patients (more than 33% of patient cohort) experienced DLT. Intra-patient dose escalation was not permitted. If a patient experienced DLT, the dose of drugs was decreased by one level at the start of the next treatment cycle.
DLTs were defined as grade 4 neutropenia for more than 5 days, grade 3 or 4 neutropenia with fever ≥38.5°C, grade 4 thrombocytopenia of any duration, grade 3 or 4 non-hematologic toxicities not improving to at least grade 1 within 2 days after institution of appropriate therapy (excluding alopecia and non-premedicated nausea and vomiting), a toxicity causing discontinuation of capecitabine or sorafenib for more than 25% of the scheduled dosage within a cycle (3 weeks), or a delay of >14 days in initiating the second cycle of therapy due to the persistent toxicity of grade 2 or higher in cycle 1. DLTs were evaluated only in the first cycle of treatment.
Dose modification
A patient could begin the next treatment cycle if ANC ≥ 1,000/mm3 and platelets ≥100,000/mm3. Below these levels, capecitabine and cisplatin were delayed and sorafenib was either administered at a decreased dose (1,000/mm3 > ANC ≥ 500/mm3) or delayed (ANC < 500/mm3) until recovery. If 1,500/mm3 > ANC ≥ 1,000/mm3 and platelets ≥100,000/mm3 at the start of the next cycle, dose reductions (25%) of capecitabine and cisplatin were made. If at any time a patient experienced serious hematologic toxicities defined as DLTs, stepwise dose reductions of capecitabine and cisplatin were made at the restart of treatment with a maximum of 50% as specified in the protocol. Capecitabine was interrupted for non-hematologic toxicities of grade 2 or worse, and an appropriate treatment was started, if possible. Missed doses were not replaced and capecitabine was restarted when the toxicities resolved or decreased in intensity to grade 1 at a reduced dose down to 50%, as specified in the protocol. Dose modification of cisplatin was done if decreased creatinine clearance, nausea/vomiting, or neurotoxicity occurred. For grade 2 or worse skin toxicities (rash/desquamation and hand-foot syndrome[HFS]), sorafenib was interrupted and restarted when the toxicity resolved or improved to grade 1 at the original dose (for the first appearance of grade 2 toxicities) or at a reduced dose (for grade 3 toxicities or repeated appearance of grade 2 toxicities).
Response and toxicity assessment
Each patient’s initial evaluation included medical history, physical examination, ECOG performance status, tumor assessment using CT scans or MRI of abdomen and pelvis, complete differential blood counts (CBC), serum chemistry and electrolytes, coagulation battery, urinalysis, electrocardiogram, chest X-ray, and pregnancy test, if indicated. Tumor response was assessed every two cycles by RECIST using the same imaging techniques and methods used at baseline. Complete and partial responses (PR) were confirmed at least 4 weeks following the initial documentation of the objective response. CBC was repeated every week while patients were on the combination therapy (up to 8 cycles). Prior to each cycle of chemotherapy, the following procedures were performed: Physical examination, performance status assessment, collection of adverse events, chemistry, electrolytes, and chest X-ray. Adverse events, oral drug administration compliance, and abnormal laboratory findings were collected at every regular visit. Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0.
Results
Patient characteristics and disposition
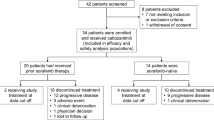
Between Oct 31, 2007, and Jul 14, 2008, 21 patients were enrolled in the study; baseline patient characteristics are listed in Table 1. Seventeen patients presented with metastatic disease initially. Two patients had received curative resections 6 and 11 months before relapses. Another two patients received surgery with curative intent, but metastatic lymph nodes and a lesion on the transverse mesocolon were grossly unresectable.
A total of 133 cycles of combination therapy were delivered (range = 1–8, median = 8) and, after completion of 8 cycles of combination therapy, a total of 82 cycles (range = 1–16, median = 9) of sorafenib monotherapy was administered to 10 patients. All patients completed at least one cycle of combination therapy and 20 patients had response evaluations after cycle 2.
As of the February 25, 2010 data cut-off date, all 21 patients had discontinued study treatment. Fifteen discontinued because of disease progression, 1 discontinued for palliative surgery after 17 cycles, 1 discontinued due to poor tolerability, and 4 discontinued due to adverse events or dose-limiting toxicities. As of the cut-off date, 14 of the 21 patients had died; 1 during the study and the rest during follow-up.
A summary of the disease characteristics and treatment results are shown in Table 2.
Dose-limiting toxicity
No DLT was observed at dose Level 1 (n = 3). One DLT (grade 3 diarrhea) was noted at dose Level 2 (n = 6), and 2 DLTs (grade 4 neutropenias lasting more than 5 days) were observed at dose Level 3 (n = 6), which made Level 3 the MTD. However, in cycle 2 and thereafter at dose Levels 2 and 3, the relative dose intensities (RDIs, the percentage of actually administered dose to planned dose per unit time) were not well maintained (mostly below 80%) due to frequent dose reductions and cycle delays (Fig. 1. Number of patients received more than 6 cycles were largely different among groups and were not shown). At dose Level 2, dose reductions were made in 66.7% of cycles and 61.9% of cycles were delayed more than 5 days. At dose Level 3, dose reductions and cycle delays were made in 90.6% and 59.4%, respectively, of all cycles. The reasons for the dose reductions and cycle delays were hematologic (about 50% of cases) and non-hematologic (about 50% of cases) toxicities at dose Level 2, while the reasons were mostly hematologic toxicities (80% of cases) at dose Level 3.

The median relative dose intensity (%) of (a) capecitabine, (b) cisplatin, and (c) sorafenib according to the dose levels and cycles
We explored a new dose level (1A) between dose Levels 1 and 2. Since no DLT was found in the 6 patients at the dose Level 1A and the RDI was mostly above 80% throughout the treatment period, Level 1A was determined as the recommended dose (RD) for the next step in clinical development. Dose reductions and cycle delays were made in 21.2% and 18.2%, respectively, of all cycles at dose Level 1A.
Safety
All 21 patients were assessable for toxicity and worst toxicities per patient are summarized in Table 3. Grade 3 and 4 neutropenia and thrombocytopenia were observed in 66.7% (14 out of 21) and 19.0% (4/21) of patients, respectively. There was only one episode of febrile neutropenia accompanied with grade 3 HFS which was treated effectively with conservative management. Grade 4 neutropenia lasting longer than 5 days was the cause of DLT in 2 patients. The most common non-hematologic toxicities of any grade were asthenia (95.2%), sensory neuropathy (85.7%), HFS (80.1%), anorexia (76.2%), nausea (66.7%), stomatitis (61.9%), and diarrhea (61.9%). Grade 3 and 4 non-hematologic toxicities were relatively uncommon. Grade 3 HFS was observed in 3 patients (14.3%) and grade 3 diarrhea and AST/ALT elevation was seen in one patient.
After completion of the scheduled 8 cycles of combination therapy, 10 patients maintained sorafenib monotherapy. Median compliance of sorafenib treatment was 100% (range 47.6–100.0%). No grade 3 or 4 toxicity was observed during sorafenib monotherapy except 2 cases of grade 3 anemia. Non-hematologic toxicities were also generally mild and were all grade 1, except one patient with grade 2 asthenia and stomatitis. HFS was reported in 3 patients (grade 1) but only in the first cycle of monotherapy; it resolved and did not reappear during maintenance.
There were 5 serious adverse events (SAEs) reported in 4 subjects. One grade 3 diarrhea was judged to be possibly related to the study drug. One patient had an episode of febrile neutropenia which was thought to be possibly related to the study drug. This patient was diagnosed later with deep vein thrombosis in a lower extremity which was managed with warfarin maintenance. Another patient had gastric perforation after cycle 2 which was fatal. A thromboembolism involving right iliac artery was reported in one patient which was possibly related to the study drug; this patient underwent embolectomy.
Efficacy
Among 16 patients with measurable disease according to RECIST criteria, 10 patients showed confirmed objective responses (all PRs); the response rate was 62.5% (95% confidence interval [CI] = 38.8–86.2). The median response duration in the 10 patients with objective response was 262 days (range 102–498). The median progression-free survival (PFS) and overall survival (OS) were 10.0 months (95% CI = 7.4–13.8) and 14.7 months (95% CI = 12.0–20.0), respectively, with 7 patients alive with a median follow-up of 23.0 months (Fig. 2).

Kaplan-Meier curve for progression-free survival (PFS, dotted line) and overall survival (OS, solid line). Median PFS and OS was 10.0 months (95% CI, 7.4–13.8) and 14.7 months (95% CI, 12.0–20.0)
Discussion
Palliative chemotherapy in patients with advanced gastric cancer improves quality of life and survival compared with best supportive care [11, 27] and has shown response rates of 25 to 50% and survival ranging 8 to 12 months [4, 16, 36]. These results have not changed with the introduction of newer agents such as taxane and irinotecan or by treating patients with three drug combinations which induce more toxicities [4, 5, 21, 36].
Sorafenib is an orally administered inhibitor of multiple kinases including RAF, MAPK, and VEGFR. Sorafenib has shown efficacy in hepatocellular and renal cell carcinoma as a monotherapy and is being investigated in various tumor types in combination with other chemotherapeutic agents. We examined sorafenib in combination with capecitabine and cisplatin (XP regimen) to determine the DLT and MTD of this combination and to evaluate the safety profile.
We defined MTD as a dose at which two or more out of six patients (more than 33% of patient cohort) experience DLTs. The recommended dose (RD) for the next step in clinical development was defined as the dose level just below the MTD dose. However, the RDIs were not maintained (mostly below 80%) at dose Levels 2 and 3 as patients continued study treatment beyond the first cycle. The usual way of determining MTD is based on the observation of toxicities in the first cycle of each patient at each dose level. The RD for further clinical development is then also being selected from the observations of first cycles. This approach is taken to shorten clinical development time and is based on the assumption that the safety profile of the first cycle is representative of the whole treatment period. The combination of sorafenib with XP showed a gradual decrease of RDIs due to frequent dose reductions, schedule delays, and poor compliance as patients continued treatment even at dose Level 2 where only one DLT was observed. Therefore, we defined a new dose level (1A) as the RD for further development to keep RDIs above 80%. Relatively big differences of RDIs were observed between dose levels with small changes of chemotherapeutic agents or sorafenib dose. It is possible that the addition of sorafenib with mild hematologic toxicities to cytotoxic chemotherapeutic agents potentiated the hematologic toxicities of this combination considering that sorafenib monotherapy caused neutropenia of all grades in 18% and grade 3 and 4 neutropenia in 5% of in renal cell carcinoma patients [9]. Also frequent dose reductions due to mild neutropenia (grade 2) in subsequent cycles according the predefined dose modification scheme contributed in lowering RDI. Judging from the difference of cisplatin dose and schedule in FP regiment between study ML17032 (80 mg/m2 every 3 weeks) and V325 (100 mg/m2 every 4 weeks) and its impact on the hematologic toxicities (19% versus 57% in grade 3 and 4 neutropenia incidence), 20 mg/m2 decrease of cisplatin dose in level 1A might have contributed in improving RDI [16, 36]. As a result, 5 patients in dose level 2 (except one patient with DLT in cycle 1 and dropped) received only 4–6 cycles of chemotherapy while 5 patients in level 1A completed 8 cycles of planned chemotherapy, and overall accumulated dose of capecitabine(X), cisplatin(P), and sorafenib(S) in level 2 was much lower than level 1A (X: 73,800~116,500 mg/m2 vs 87,800~174,300 mg/m2, P: 240~465 mg/m2 vs 300~460 mg/m2, S: 52,800~74,400 vs 69,200~140,000 mg).
The most common toxicities seen with sorafenib plus XP included anemia (100% of patients, any grade), thrombocytopenia (100%), asthenia (95.2%), neutropenia (90.5%), sensory neuropathy (85.7%), HFS (80.1%), and diarrhea (61.9%). The incidence of grade 3 and 4 neutropenia in the present study (66.7%, Table 3) appears to be quite high compared to 16% from ML17032 study with XP regimen in a similar patient population [16]. This difference might originate from frequent evaluation of hematologic toxicities (weekly CBC in the present study vs triweekly CBC in the ML17032 study). Considering that sorafenib monotherapy caused neutropenia of all grades in 18% and grade 3 and 4 neutropenia in 5% of in renal cell carcinoma patients, it is also possible that the addition of sorafenib amplified the hematological toxicity of XP regimen [9]. HFS and diarrhea were most common adverse events with both sorafenib and capecitabine monotherapies, with an incidence of 54–68% and 27–57% with capecitabine [2, 12, 13, 29, 34] and 21–30% and 39–43% with sorafenib [9, 20]. However, these two common toxicities seemed to be modestly increased by the combination used in the present study (80.1% and 61.9% for HFS and diarrhea, respectively) but were mostly mild or moderate in severity (Table 3). The incidence of HFS with combination therapy was lower than the arithmetical sum of incidences seen in the studies of capecitabine and sorafenib monotherapies which might be due to the difference of the proposed mechanism by which capecitabine and sorafenib induce HFS.
Among the 5 SAEs reported, there was one deep vein thrombosis of a lower extremity and one arterial thromboembolism. Vascular thromboembolic and hemorrhagic adverse events of antiangiogenic therapies have been widely reported and vary depending on the mechanism of the drugs administered, the combinations with chemotherapy used, and the type of tumor being treated [7]. Although thromboembolism has not been prevalent in previous clinical trials of sorafenib monotherapy, thromboembolic and bleeding complications should be carefully monitored in future studies employing combination chemotherapies since SU5416, a VEGFR-1/-2 inhibitor, showed high thrombotic potential only when combined with other therapeutic agents [3, 18].
Perforation of a gastric ulcerative lesion at the antrum and resulting peritonitis and death occurred in 1 patient in the present study. This event developed after cycle 2 and was associated with a 37% reduction of measurable tumor lesions assessed by RECIST criteria. Although some gastrointestinal perforations were reported in sorafenib-treated patients without gastrointestinal lesions [8, 25], it is uncertain whether the perforation in the patient in the present study was influenced by the antiangiogenic activity of sorafenib or was a consequence of an antitumor effect in an ulcerative, already perforation-prone lesion. In any event, patients with ulcerative lesions who could respond to the treatment with a perforation of primary gastric tumor should be carefully monitored in future clinical trials of sorafenib with or without cytotoxic chemotherapy.
Sorafenib was also tested in advanced gastric cancer patients in combination with docetaxel and cisplatin in a phase II trial. [33] Without precedent dose-finding study, full dose of sorafenib (400 mg bid) was combined with full dose of docetaxel (75 mg/m2) and cisplatin (75 mg/m2) combination. The doses of agents in this phase II study seem to be too high to be tolerated, and 64% of patients experienced grade 3 and 4 neutropenia and 23% of patients had their treatment terminated before four cycles of therapy because of chemotherapy-related toxicity. Another 23% of patients had either cisplatin or docetaxel dose reductions to 75%, and one patient had both medications reduced to 50% of initial dose. Of the 44 patients, 35 had their sorafenib dose held or modified per protocol at some point during treatment. Nineteen patients needed a sorafenib dose interruption or modification during the first cycle and 11 patients required two or more such sorafenib modifications. Despite the toxicities and poor tolerability, the efficacy results (ORR 41%, PFS 5.8 months, and OS 13.6 months) seem to be comparable but not superior to the precious results of other combination chemotherapy used in AGC. Thus the cautious dose finding study is needed considering both tolerability and maintenance of dose intensities for combination of multitarget kinase inhibitor such as sorafenib with chemotherapy agents.
Compared to ORR and OS from trials of combination chemotherapy (25–50% and 8–12 months) [4, 16, 36] and from studies of bevacizumab (38.0% and 12.1 months, 13.9 months in Asia-Pacific patients) and trastuzumab (47.3% and 13.5 months) combined with chemotherapy in AGC patients, 62.5% ORR and 14.7 months of OS are encouraging result [15, 35]. Despite the limitations such as small number of enrolled patients and inclusion of some patients with low tumor burden such as grossly incomplete resection or M1 abdomen lymph nodes as only metastatic lesion, these results together with acceptable toxicity profiles justifie further clinical development of this combination in the treatment of metastatic or unresectable gastric cancer.
In conclusion, the combination of sorafenib 400 mg p.o. bid daily with capecitabine 800 mg/m2 p.o. bid on days 1–14 and cisplatin 60 mg/m2 iv on day 1 in a 3 week cycle is a feasible regimen for further development in advanced gastric cancer
References
Barnes CJ, Bagheri-Yarmand R, Mandal M, Yang Z, Clayman GL, Hong WK, Kumar R (2003) Suppression of epidermal growth factor receptor, mitogen-activated protein kinase, and Pak1 pathways and invasiveness of human cutaneous squamous cancer cells by the tyrosine kinase inhibitor ZD1839 (Iressa). Mol Cancer Ther 2:345–351
Blum JL, Jones SE, Buzdar AU, LoRusso PM, Kuter I, Vogel C, Osterwalder B, Burger HU, Brown CS, Griffin T (1999) Multicenter phase II study of capecitabine in paclitaxel-refractory metastatic breast cancer. J Clin Oncol 17:485–493
Cooney MM, Tserng KY, Makar V, McPeak RJ, Ingalls ST, Dowlati A, Overmoyer B, McCrae K, Ksenich P, Lavertu P, Ivy P, Hoppel CL, Remick S (2005) A phase IB clinical and pharmacokinetic study of the angiogenesis inhibitor SU5416 and paclitaxel in recurrent or metastatic carcinoma of the head and neck. Cancer Chemother Pharmacol 55:295–300
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman AR (2008) Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 358:36–46
Dank M, Zaluski J, Barone C, Valvere V, Yalcin S, Peschel C, Wenczl M, Goker E, Cisar L, Wang K, Bugat R (2008) Randomized phase III study comparing irinotecan combined with 5-fluorouracil and folinic acid to cisplatin combined with 5-fluorouracil in chemotherapy naive patients with advanced adenocarcinoma of the stomach or esophagogastric junction. Ann Oncol 19:1450–1457
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Elice F, Jacoub J, Rickles FR, Falanga A, Rodeghiero F (2008) Hemostatic complications of angiogenesis inhibitors in cancer patients. Am J Hematol 83:862–870
Eng FC, Easson AM, Szentgyorgyi E, Knox JJ (2009) Sorafenib and surgical complications: a case report of adverse reaction to sorafenib during treatment for renal cell carcinoma. Eur J Surg Oncol 35:219–221
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356:125–134
Flaherty KT, Schiller J, Schuchter LM, Liu G, Tuveson DA, Redlinger M, Lathia C, Xia C, Petrenciuc O, Hingorani SR, Jacobetz MA, Van Belle PA, Elder D, Brose MS, Weber BL, Albertini MR, O’Dwyer PJ (2008) A phase I trial of the oral, multikinase inhibitor sorafenib in combination with carboplatin and paclitaxel. Clin Cancer Res 14:4836–4842
Glimelius B, Ekstrom K, Hoffman K, Graf W, Sjoden PO, Haglund U, Svensson C, Enander LK, Linne T, Sellstrom H, Heuman R (1997) Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann Oncol 8:163–168
Hoff PM, Ansari R, Batist G, Cox J, Kocha W, Kuperminc M, Maroun J, Walde D, Weaver C, Harrison E, Burger HU, Osterwalder B, Wong AO, Wong R (2001) Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J Clin Oncol 19:2282–2292
Hong YS, Song SY, Lee SI, Chung HC, Choi SH, Noh SH, Park JN, Han JY, Kang JH, Lee KS, Cho JY (2004) A phase II trial of capecitabine in previously untreated patients with advanced and/or metastatic gastric cancer. Ann Oncol 15:1344–1347
Kanai T, Konno H, Tanaka T, Matsumoto K, Baba M, Nakamura S, Baba S (1997) Effect of angiogenesis inhibitor TNP-470 on the progression of human gastric cancer xenotransplanted into nude mice. Int J Cancer 71:838–841
Kang Y, Ohtsu A, Van Cutsem E, Rha SY, Sawaki A, Park S, Lim H, Wu J, Langer B, Shah MA (2010) AVAGAST: a randomized, double-blind, placebo-controlled, phase III study of first-line capecitabine and cisplatin plus bevacizumab or placebo in patients with advanced gastric cancer (AGC). J Clin Oncol 28:(15) abstr LBA4007
Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L, Philco-Salas M, Suarez T, Santamaria J, Forster G, McCloud PI (2009) Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: a randomised phase III noninferiority trial. Ann Oncol 20:666–673
Kim TW, Kang YK, Ahn JH, Chang HM, Yook JH, Oh ST, Kim BS, Lee JS (2002) Phase II study of capecitabine plus cisplatin as first-line chemotherapy in advanced gastric cancer. Ann Oncol 13:1893–1898
Kuenen BC, Rosen L, Smit EF, Parson MR, Levi M, Ruijter R, Huisman H, Kedde MA, Noordhuis P, van der Vijgh WJ, Peters GJ, Cropp GF, Scigalla P, Hoekman K, Pinedo HM, Giaccone G (2002) Dose-finding and pharmacokinetic study of cisplatin, gemcitabine, and SU5416 in patients with solid tumors. J Clin Oncol 20:1657–1667
Kupsch P, Henning BF, Passarge K, Richly H, Wiesemann K, Hilger RA, Scheulen ME, Christensen O, Brendel E, Schwartz B, Hofstra E, Voigtmann R, Seeber S, Strumberg D (2005) Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin Colorectal Cancer 5:188–196
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359:378–390
Lokich JJ, Sonneborn H, Anderson NR, Bern MM, Coco FV, Dow E, Oliynyk P (1999) Combined paclitaxel, cisplatin, and etoposide for patients with previously untreated esophageal and gastroesophageal carcinomas. Cancer 85:2347–2351
Lordick F, Lorenzen S, Hegewisch-Becker S, Folprecht G (2007) Cetuximab plus weekly oxaliplatin/5FU/FA (FUFOX) in 1st line metastatic gastric cancer. Final results from a multicenter phase II study of the AIO upper GI study group. J Clin Oncol. p. (abstr 4526).
Magne N, Fischel JL, Dubreuil A, Formento P, Poupon MF, Laurent-Puig P, Milano G (2002) Influence of epidermal growth factor receptor (EGFR), p53 and intrinsic MAP kinase pathway status of tumour cells on the antiproliferative effect of ZD1839 (“Iressa”). Br J Cancer 86:1518–1523
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
Peters NA, Richel DJ, Verhoeff JJ, Stalpers LJ (2008) Bowel perforation after radiotherapy in a patient receiving sorafenib. J Clin Oncol 26:2405–2406
Pinto C, Di Fabio F, Siena S, Cascinu S, Rojas Llimpe FL, Ceccarelli C, Mutri V, Giannetta L, Giaquinta S, Funaioli C, Berardi R, Longobardi C, Piana E, Martoni AA (2007) Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann Oncol 18:510–517
Pyrhonen S, Kuitunen T, Nyandoto P, Kouri M (1995) Randomised comparison of fluorouracil, epidoxorubicin and methotrexate (FEMTX) plus supportive care with supportive care alone in patients with non-resectable gastric cancer. Br J Cancer 71:587–591
Richly H, Henning BF, Kupsch P, Passarge K, Grubert M, Hilger RA, Christensen O, Brendel E, Schwartz B, Ludwig M, Flashar C, Voigtmann R, Scheulen ME, Seeber S, Strumberg D (2006) Results of a Phase I trial of sorafenib (BAY 43-9006) in combination with doxorubicin in patients with refractory solid tumors. Ann Oncol 17:866–873
Sakamoto J, Chin K, Kondo K, Kojima H, Terashima M, Yamamura Y, Tsujinaka T, Hyodo I, Koizumi W (2006) Phase II study of a 4-week capecitabine regimen in advanced or recurrent gastric cancer. Anticancer Drugs 17:231–236
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Shah MA, Ramanathan RK, Ilson DH, Levnor A, D’Adamo D, O’Reilly E, Tse A, Trocola R, Schwartz L, Capanu M, Schwartz GK, Kelsen DP (2006) Multicenter phase II study of irinotecan, cisplatin, and bevacizumab in patients with metastatic gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol 24:5201–5206
Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ (2006) Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res 12:144–151
PM SW, O’Dwyer PJ, Catalano P, Ansari RH, Benson AB 3rd (2010) Phase II study of sorafenib in combination with docetaxel and cisplatin in the treatment of metastatic or advanced gastric and gastroesophageal junction adenocarcinoma: ECOG 5203. J Clin Oncol 28:2947–2951
Twelves C, Wong A, Nowacki MP, Abt M, Burris H 3rd, Carrato A, Cassidy J, Cervantes A, Fagerberg J, Georgoulias V, Husseini F, Jodrell D, Koralewski P, Kroning H, Maroun J, Marschner N, McKendrick J, Pawlicki M, Rosso R, Schuller J, Seitz JF, Stabuc B, Tujakowski J, Van Hazel G, Zaluski J, Scheithauer W (2005) Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 352:2696–2704
Van Cutsem E, Kang Y, Chung H, Shen L, Sawaki A, Lordick F, Hill J, Lehle M, Feyereislova A, Bang Y (2009) Efficacy results from the ToGA trial: a phase III study of trastuzumab added to standard chemotherapy (CT) in first-line human epidermal growth factor receptor 2 (HER2)-positive advanced gastric cancer (GC). J Clin Oncol 27:abstr LBA4509
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, Risse ML, Ajani JA (2006) Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24:4991–4997
Yoshikawa T, Tsuburaya A, Kobayashi O, Sairenji M, Motohashi H, Yanoma S, Noguchi Y (2000) Plasma concentrations of VEGF and bFGF in patients with gastric carcinoma. Cancer Lett 153:7–12
Acknowledgement
This work was supported by the donation of study drug (sorafenib) from Bayer Schering Pharma AG. Part of this work was presented as a poster at the 45th ASCO annual meeting in Orlando, Florida, USA and the ECCO 15 and 34th ESMO multidisciplinary congress in Berlin, Germany, 2009.
Financial disclosure
Dr Yoon-Koo Kang received honoraria for lecture and advisory board from Bayer Schering Pharma AG and Roche. Besides, there is no conflict of interest to be reported from other authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kim, C., Lee, JL., Choi, Y.H. et al. Phase I dose-finding study of sorafenib in combination with capecitabine and cisplatin as a first-line treatment in patients with advanced gastric cancer. Invest New Drugs 30, 306–315 (2012). https://doi.org/10.1007/s10637-010-9531-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-010-9531-2