
Summary
Cumulative evidence has established that hypoxia-inducible factor-1α (HIF-1α) and its downstream target, vascular endothelial growth factor (VEGF), play a critical role in hepatocellular carcinoma angiogenesis, invasiveness and metastasis. 3-(4-bromophenyl)-2-(ethylsulfonyl)-6-methylquinoxaline 1,4-dioxide (Q39) has recently shown great antiproliferative activity in extensive cell lines in normoxia and hypoxia. In this study, Q39 exhibited high antiproliferative activity against hepatoma both in vitro and in vivo, mainly by inducing apoptosis. In addition, suppression of HIF-1α by Q39 resulted in a drastic decrease in VEGF expression. These results indicate that Q39 is an effective inhibitor of HIF-1α and provide new perspectives into the mechanism of its anticancer activity. Interestingly, neither the HIF-1α degradation rate nor the HIF-1α steady-state mRNA level was affected by Q39. Instead, suppression of HIF-1α accumulation by Q39 correlated with prominent dephosphorylation of mTOR and 4E-BP1, a pathway known to regulate protein expression at the translational level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypoxic areas in solid tumors are more resistant to chemotherapy and radiotherapy than aerobic ones. However, hypoxia can also be used to produce a novel class of specific anti-tumor prodrugs, namely bioreductive agents, including quinones, nitro derivatives, and N-oxides.
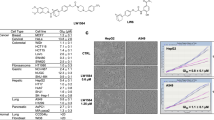
Q39, 3-(4-Bromophenyl)-2-(ethylsulfonyl)-6-methylquinoxaline 1,4-dioxide, was initially discovered in the Hu laboratory (Fig. 1a). Previously, our lab has shown using a cytotoxicity assay that Q39 can inhibit the proliferation of several types of tumors such as leukemia, prostate, esophagus, liver and stomach cancers. Q39 showed remarkable hypoxic selectivity in K562, PC3 and SMMC-7721 cells. Our studies also showed that Q39 can inhibit murine S180 and H22 bearing mice in vivo (unpublished data). Flow cytometry and immunostaining assays demonstrated that Q39 induced apoptosis in K562 cells in hypoxia, and the mechanism seemed to involve multiple apoptotic pathways, including the regulation of mitochondrial function and enhancement of caspase-9/3 expression and activation. Most importantly, Q39 inhibited hypoxia-inducible factor 1 α (HIF-1α) accumulation and decreased the expression of vascular endothelial growth factor (VEGF) in K562 cells [1, 2].

The hypoxic selective activity of Q39 correlates with expression of HIF-1α. a The chemical structure of Q39. b Hepatocellular carcinoma cells (SMMC-7721 and HepG2) were seeded onto 96-well plates (5 × 103 per well) for 24 h and subsequently treated with different concentrations of Q39 (0–40 μM) under normoxia or hypoxia for 72 h. Cell viability was assayed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method. Representative of three independent experiments. c Hepatocellular carcinoma cells were incubated in 20% O2 or 3% O2 for 24 h. Whole-cell lysates were collected and analyzed by immunoblotting for expression of HIF-1α
As the master regulator of the hypoxic transcriptional response, HIF-1 plays a central role in tumor growth and angiogenesis [3]. HIF-1 is a heterodimeric transcription factor composed of HIF-1α and HIF-1β subunits. Under hypoxic conditions, due to blockage of prolyl hydroxylation, HIF-1α escapes from 26 S proteasome system degradation, translocates, accumulates in the nucleus rapidly, and forms a heterodimer with HIF-1β. This process results in the up-regulation of an array of genes involved in angiogenesis, erythropoiesis, energy metabolism and cell survival [4]. Vascular endothelial growth factor (VEGF), a downstream gene of HIF-1, is one of many principal mediators of tumor angiogenesis [5]. Inhibition of VEGF does not only block cancer cell proliferation in vitro and tumor growth in vivo [6, 7] but also contributes to the decrease of cancer growth, development and metastasis [8]. HIF-1 and VEGF activation require the recruitment of multiple signaling components, including ERK1/2 and PI3K/AKT/mTOR/4E-BP1. The PI3K/AKT/mTOR/4E-BP1 signaling pathway appears to positively regulate HIF-1 and VEGF activation. Notably, recent research on PI3K/AKT/mTOR/4E-BP1 has shown that the pathway is constitutively activated in many tumors, suggesting that targeting this pathway can directly or indirectly inhibit tumor growth and progression.
In this study, we have shown for the first time that Q39 suppresses hypoxia-induced HIF-1α and VEGF expression in human hepatoma (SMMC-7721) cells but has no apparent inhibitory effects on HIF-1α mRNA levels. The suppression of HIF-1α correlated with blockage of HIF-1α protein accumulation via Akt/mTOR/4E-BP1 signaling pathways but not by increasing HIF-1α protein degradation. We believe Q39 triggered dephosphorylation of 4E-BP1, which interrupted the interaction between 4E-BP1 and eIF4E. These unique findings provide a further understanding of the molecular mechanisms underlying the anti-angiogenic effects of Q39 and in turn, help to delineate a novel means for regulation of protein expression and define the regulation of HIF-1α at the translational level.
Materials and methods
Reagents
Q39 was synthesized by prof. Hu, which was reported in our previous study [1]. Stock solution of Q39 (20 mM) was prepared with dimethyl sulfoxide (DMSO) and stored at −20°C for in vitro test. The stock solution was further diluted with the appropriate assay medium immediately before use. LY294002 (Calbiochem, San Diego, CA) was reconstituted in DMSO. The final DMSO concentration did not exceed 0.1% throughout the study. The selective proteasome inhibitor, MG132 (Z-Leu-Leu-Leu-CHO), and protein synthesis inhibitor, cycloheximide (CHX), were from EMD Biosciences, Inc. (La Jolla, CA) and Sigma (St. Louis, MO) respectively. Antibodies for HIF-1α, VEGF, eIF4E, p-4EBP1, β-actin or AKT and β-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for 4EBP1, mTOR, p-mTOR, AKT (Ser473) were from Cell Signaling Technology, Inc., (Beverly, MA).
Cell culture and establishment of hypoxia culture condition
Hepatocarcinoma cell lines (SMMC-7721 and HepG2) were purchased from Cell Bank of China Science Academy (Shanghai, China). Hepatoma cells were maintained in RPMI 1640 or DMEM medium (Invitrogen Corp., Carlsbad, CA) plus 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 units/mL) and streptomycin (100 μg/mL) and incubated at 37°C in a humidified atmosphere with 5% CO2. Moderate hypoxic conditions (3% O2) were established in a hypoxia incubator (Forma Scientific, Inc., Marietta, OH) where N2 was used to compensate for the reduced O2 level.
Cell viability assay and apoptosis assay
SMMC-7721 or HepG2 cells were plated onto 96-well plates (5 × 103 per well) for 24 h and subsequently treated with different concentrations of Q39 under normoxia or hypoxia for 72 h. Viable cells were determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Assay kit (Sigma) according to the manufacturer’s instructions. Apoptosis of SMMC-7721 cells caused by Q39 were analyzed by Annexin V-FITC/Propidium iodide (PI) apoptosis detection kit (BioVision, Mountain View, CA) and flow cytometry as described [2]. Briefly, cells (3 × 105 per sample) were washed in PBS twice and resuspended in 500 μL of binding buffer plus Annexin V-FITC and Propidium iodide. Following incubation for 15 min in the dark, cell nuclei were analyzed with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). DNA content of the nuclei was registered on a logarithmic scale.
Real-time PCR analysis for HIF-1α and VEGF mRNA levels
Total RNA from SMMC-7721 cells (1 × 106) was extracted by Trizol reagent (Bio Basic Inc., Markham, Ontario, Canada) and cDNA was synthesized using 2 µg of total RNA with random hexamer primers and RevertAidTM M-MuLV Reverse Transcriptase (Fermentas International Inc., Burlington, Ontario, Canada). The conditions used for Reverse Transcription-PCR were as follows: 10 min at 25°C, 60 min at 48°C, 15 min at 72°C and 10 min at 95°C. Typically 100 ng of reverse-transcribed cDNA per sample were used to perform real-time PCR analysis of HIF-1α, VEGF and GAPDH mRNA levels which was analyzed by QuantiTectTM SYBR Green PCR kits (Qiagen Inc., Valencia, CA). The housekeeping gene GAPDH was used as an internal standard. The Real-Time PCR protocol consisted of thermal cycling as follows: initial denaturation at 95°C for 15 min, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s using an Eppendorf epGradient Mastercycler (Eppendorf, Hamburg, Germany). The following primers used for the PCR were as follows: HIF-1α, forward prime 5′-TCACCACAGGACAGTACAGGATGC-3′ and reverse prime 5′-CCAGCAAAGTTAAAGCATCAGGTTCC-3′; VEGF, forward prime 5′-AGGAGGGCAGAATCATCACG-3′ and reverse prime 5′-CAAGGCCCACAGGGATTTTCT-3′; GAPDH, forward prime 5′-GTCATCCATGACAACTTTGG-3′and reverse prime 5′-GAGCTTGACAAAGTGGTCGT-3′. In all experiments, two negative controls were carried through all steps. Values are expressed as fold increases relative to the reference sample (medium).
Western blot analysis
The protein samples from SMMC-7721 cells were prepared as described previously [2]. Protein extracts were resolved by 8%–15% SDS-PAGE and electroblotted onto PVDF membranes, and then western blot analysis were carried out using specific primary antibodies following by incubation with a horseradish peroxidase–conjugated secondary antibody (1:5000). The signals were visualized by the enhanced chemiluminescence detection system (Biological Industries, Beit Haemek, Israel). As a loading control, the blots were reprobed with a specific antibody against human α-tubulin or β-actin (1:1000).
Immunoprecipitation
Cells were harvested and lysed using universal lysis/immunoprecipitation buffer (50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 25 mM NaFl, 25 mM β-glycerophosphate pH 7.5, 0.1 mM sodium orthovanadate, 0.1 mM PMSF, 5 µg of leupeptin per ml, 0.2% Triton X-100, 0.5% Nonidet P-40). The protein concentration was measured by the Bradford method (Bio-Rad). Aliquots (500 µg) of cellular proteins were precleaned by adding 1.0 µg of the appropriate normal immunoglobulin G together with 20 µl of appropriate protein A + G-agarose conjugate (Santa Cruz) for 1 h at 4°C. The immunoprecipitations were performed with the appropriate antibody for 2 to 16 h at 4°C. Complexes bound to the protein A + G-agarose conjugate were washed five times with universal lysis/immunoprecipitation buffer and separated by SDS-PAGE. The Western blotting was performed as described above.
Transient transfection
Chemically synthesized double-stranded siRNA specific for HIF-1α (siRNAHIF-1α), 5′- CUGAUGACCAGCAACUUGATT-3′ and 5′-UCAAGUUGCUGGUCAUCAGTT-3′, was purchased from GenePharma Company (Shanghai, China). The siRNA was transfected (40.0 nM) using lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. A non-targeting siRNA sequence (GenePharma) was used as nonspecific control.
ELISA for VEGF production
VEGF production in the conditioned medium was assayed with Quantikine Human VEGF ELISA kit (R&D Systems, Inc., Minneapolis, MN) according to the manufacturer’s protocols. Results were normalized to cell count.
Chick embryo chorioallantoic membrane (CAM) assay
Inhibition of angiogenesis was determined using a modification of the CAM assay. Fertilized chick eggs were stayed in an egg incubator regulated at 37°C and with 50% humidity for optimal growth conditions for 6 days continuously. Then the egg shell was cracked, and gently opened into the plate to avoid any unnecessary physical stress. It was made sure that the yolk sac membrane remained intact and that the embryo was viable. A good embryo with a beating heart was chosen, and the rest were discarded. The sterile filter paper square saturated with Q39 (2 and 8 μg) or physiological saline solution was placed in areas between vessels but never onto any large vessels. After 48 h, the CAMs were carefully isolated and were fixed in methanol/acetone. Results of the samples were photographed.
Measurement of in vivo activity
Tumors were established by injection of SMMC-7721 cells (5 × 106 cells per animal, subcutaneously into the armpit of the athymic mice) to 5–6-week-old BALB/c male athymic mice (National Rodent Laboratory Animal Resource, Shanghai Branch, China). Treatments were initiated when tumors reached a mean group size of about 50 mm3. Tumor volume (mm3) was measured with calipers and calculated as (W2 × L)/2, where W is the width and L is the length. The T/C% was determined by calculating relative tumor volume (RTV) as T/C%=100 × (mean RTV of treated group)/(mean RTV of control group). The tumor growth inhibition rate was calculated by using the formula IR (%) = (1-TWt/TWc) × 100, where TWt and TWc were the mean tumor weight of treatment and control groups. Athymic mice was administrated intravenously with Q39 (10.0 and 2.5 mg/kg) dissolved in cremophor: ethanol: 0.9% sterile sodium chloride solution (1: 1: 8, volume) once a day. Mice weight and tumor volume were recorded every 3 days until animals were sacrificed. Animal care was in accordance with institutional guidelines.
Immunohistochemistry
Immunohistochemistry was performed as described previously. Paraffin sections (5 µm thick) of SMMC-7721 tumor were incubated with anti-HIF-1α or anti-VEGF (Santa Cruz, CA),diluted 1:100, 4°C overnight and biotin-labeled mouse IgG for 30 min followed with streptavidin-HRP for 20 min at room temperature. Lastly, sections were developed with 3,30-diaminobenzidine and counter stained with hematoxylin.
Statistical analysis
Data are presented as mean ± SD for three separate experiments in vitro and SE for three separate experiments in vivo. Comparisons between groups were made with unpaired Student’s two-tailed t test and P < 0.05 was considered statistically significant.
Results
Q39 inhibits hepatoma cell proliferation both in vitro and in vivo through induction of apoptosis in SMMC-7721 cells
To determine the effect of Q39 on hepatoma cell proliferation, SMMC-7721 and HepG2 hepatoma cells were plated onto 96-well plates (5 × 103 cells per well) for 24 h and subsequently treated with fresh medium with or without various concentrations of Q39 under normoxia or hypoxia for 72 h. All hepatoma cells exhibited dose-dependent sensitivity to Q39 both in hypoxia and normoxia (Fig. 1b). Importantly, Q39 displayed similar hypoxic toxicity in both hepatoma cell lines. However, Q39 expressed different hypoxic selective activity (normoxia/hypoxia) in SMMC-7721 and HepG2 (Fig. 1b). Additionally, our previous results suggested that the hypoxic selective activity of Q39 is not seen in all cell lines [1, 2]. These results are consistent with the fact that Q39 was originally designed as a hypoxia selective cytotoxin.
Numerous lines of evidence suggest that HIF-1, as the master regulator of the hypoxic transcriptional response, plays a crucial role in tumor growth and angiogenesis [3]. Moreover, many studies have indicated that HIF-1α and its downstream target, VEGF, regulate angiogenesis and play an important role in hepatocellular carcinoma (HCC) growth [9]. Here, we attempt to unravel the relationship between HIF-1α protein expression and the anti-proliferative activity of Q39. As shown in Fig. 1c, in SMMC-7721 cells, HIF-1α protein expression increased under hypoxic conditions (Fig. 1c), which correlated with the hypoxic selective activity of Q39. Interestingly, there was no change of HIF-1α expression in HepG2 cells under the same conditions (Fig. 1c). Of note was the fact that the basal level of HIF-1α expression in HepG2 cells was the same at different oxygen concentrations (Fig. 1c). These observations in HepG2 cells might explain the comparable anti-proliferation activity of Q39 under both hypoxia and normoxia in these cells.
Given the hypoxia selectivity of Q39, we chose the SMMC-7721 cell line for further analysis. To study whether the decrease in cell proliferation was due to Q39-elicited apoptosis, we determined the apoptotic population in Q39-treated SMMC-7721 cells using Annexin V/PI staining and flow cytometry. The percentage of apoptotic cells (Annexin V+) increased from 9.1% to 80.1% in a dose-dependent manner in hypoxia (Fig. 2a, b). These results indicate that Q39 induces apoptosis in SMMC-7721 cells.

Q39 inhibits hepatoma cell proliferation through induction of apoptosis in SMMC-7721 cells under hypoxic condition. a SMMC-7721 cells were cultured to 80% confluence and then exposed to hypoxia with DMSO, 2.5, 5, and 10 μM Q39 for 48 h. Cells were also exposed to normoxia with DMSO. Cells were then trypsinized, harvested, washed twice with PBS buffer, and stained using Annexin V-FITC/Propidium iodide (PI). Stained cells were then subjected to flow cytometry assay by a FACSCalibur system (BD Biosciences) to analyze the apoptotic cells. b Quantitative analysis of results from C. Columns: mean percentage of apoptotic cells from three replicate experiments; bars: SE. c Q39 potently inhibited tumor growth in a SMMC-7721 xenograft tumor model
Although cell-based experiments demonstrated the anti-proliferative action of Q39 in vitro, the in vivo anti-hepatoma activity of Q39 remained to be elucidated. The in vivo anti-tumor activity of Q39 was evaluated using human tumor models xenografted in athymic mice. Q39 inhibited SMMC-7721 tumor growth in a dose-dependent manner (Fig. 2c). Compared to the control group, Q39 significantly effected tumor weight, as indicated by the tumor growth inhibition rates of 66.7% (p < 0.01) in the 10.0 mg/kg group and 53.3% (p < 0.05) in the 2.5 mg/kg group. The T/C% was 27.2 (p < 0.001) and 42.9 (p < 0.001) in the groups treated with Q39 (10.0 mg/kg and 2.5 mg/kg, respectively). Furthermore, there was no significant change in athymic mice body weight during the experiment, which may be evidence that the anti-hepatoma activity of Q39 takes precedence over toxicity in athymic mice. Consequently, Q39-treatment in vivo was a tolerable therapeutic method in treating SMMC-7721 cancer cells.
Q39 inhibits hypoxia-induced HIF-1α and VEGF expression in SMMC-7721 cells
Considering the importance of HIF-1α in hypoxia and its potential correlation with Q39 activity, we were prompted to test the effect of Q39 on HIF-1α expression in hepatoma cells. Exponentially growing cells (70–80% confluence) in complete medium were treated with different concentrations of Q39 and incubated under normoxic or hypoxic conditions for 16 h. Proteins were extracted from the SMMC-7721 cells, and HIF-1α was detected by western blot analysis. Our results show that pretreatment of SMMC-7721 cells with Q39 (0–20 μM) significantly inhibited hypoxia-induced HIF-1α protein accumulation in a dose-dependent manner (Fig. 3a, b). To determine whether the reduction of hypoxia-induced HIF-1α protein accumulation by Q39 was the result of transcriptional inhibition, HIF-1α mRNA levels were determined by real-time PCR under the same exposure period. As shown in Fig. 3c, treatment of SMMC-7721 cells with different concentrations of Q39 did not alter the HIF-1α mRNA levels under both normoxic and hypoxic conditions. These results suggest Q39 does not suppress hypoxia-induced HIF-1α protein accumulation at the transcriptional level. To further confirm the effect of Q39 on hypoxia-induced VEGF expression, an immediate downstream target gene of HIF-1α, the protein expression and mRNA levels of VEGF were determined by western blotting and real-time PCR in Q39-treated SMMC-7721 cells. Our results indicate that a notable increase of VEGF protein and mRNA were observed after exposure to hypoxia for 16 h. Pretreatment of Q39 significantly suppressed hypoxia-induced VEGF protein and mRNA levels in a dose-dependent manner under hypoxia (Fig. 3d–f). Additionally, both immunohistochemistry of tumor tissue in athymic mice treated with Q39 (Fig. 3g) and chick embryo chorioallantoic membrane assays (Fig. S1) indicated that Q39 blocked the production of HIF-1α and its downstream VEGF in tumor cells and exhibited potential antiangiogenic activities.

Q39 inhibits hypoxia-induced HIF-1α protein accumulation and VEGF expression in SMMC-7721 cells. a, c, d and e SMMC-7721 cells were pretreated for 1 h with different concentrations of Q39 (0–10 μM) in normal culturing conditions followed by exposure to normoxia (21% O2) or hypoxia (3% O2) for 16 h. HIF-1α/VEGF protein and mRNA levels were determined by western blot analysis (a and d) and real time-PCR (c and e), respectively. The relative fold changes of VEGF mRNA compared to GAPDH with nontreatment under normoxia was arbitrarily set as 1.0. Columns: mean of three replicate experiments; bars: SD. b densitometry analyses of A. The relative density ratio of HIF-1α/VEGF protein band to α-tubulin with nontreatment under hypoxia/normoxia was arbitrarily set as 1.0. f SMMC-7721 cells were pretreated with or without different concentrations of Q39 (0–10 μM) for 1 h in normal conditions followed by exposure to normoxia or hypoxia (3% O2) for 16 h. VEGF protein production in the conditioned medium was determined by ELISA. g Effect of Q39 against primary tumor expression of HIF-1α and its downstream VEGF. Typical photograph of primary tumor sections with immunohistochemistry staining (magnification x200)
In order to examine whether the effect of Q39 on the expression of VEGF was dependent on HIF-1α, we determined the effect of Q39 with and without siRNA transfection of HIF-1α on VEGF transcriptional activation and examined protein expression in cells by real time PCR, ELISA, and western blot analysis. Q39 decreased hypoxia-induced VEGF protein expression, mRNA levels and VEGF protein production in a dose-dependent manner in SMMC-7721 cells (Fig. 3d–f). SMMC-7721 cells were transfected with HIF-1α siRNA or nonspecific siRNA and subsequently exposed to hypoxia for 16 h with or without 20 μM Q39. The results show that expression of HIF-1α was inhibited by both Q39 and siRNAHIF-1α (Fig. 4a, b). Both siRNA of HIF-1α and Q39 potently reversed the hypoxia-induced VEGF protein production and mRNA levels (Fig. 4c, d). Compared to Q39 or siRNA of HIF-1α, Q39 plus siRNA of HIF-1α did not alter VEGF protein production and mRNA levels significantly (Fig. 4c, d), which was consistent with the change in HIF-1α protein expression (Fig. 4b, last three lanes). Taken together, these results suggest that HIF-1α contributes to Q39-induced reduction in VEGF expression, rather than Q39-driven direct regulation of VEGF expression in SMMC-7721 cells.

Q39 Inhibits Hypoxia-Induced VEGF Through the HIF-1 Pathway. a, b, c and d SMMC-7721 cells were transfected with siRNAHIF-1α or nonspecific siRNA for 4 h and subsequently exposed to normoxia or hypoxia for 16 h with (b, c, d) or without (a) 10 μM Q39. HIF-1α protein levels were analyzed by western blot (a and b). VEGF mRNA levels and VEGF protein production in the conditioned medium were determined by real time-PCR (c) and ELISA (d). Columns: mean of three independent experiments; bars, SD
Q39 inhibits hypoxia-induced HIF-1α protein expression by decreasing its synthesis but not by promoting its degradation
It has been well established that HIF-1α is regulated by many compounds through both protein degradation and protein synthesis [10–13]. To explore whether Q39 effects hypoxia-induced stabilization of HIF-1α, we exposed SMMC-7721 cells to hypoxia for 16 h followed by treatment with cycloheximide (CHX) to block ongoing protein synthesis in the presence or absence of Q39 at different time points. Our results show that Q39 slightly promoted the degradation of hypoxia-induced HIF-1α protein (Fig. 5b) compared to cells treated with CHX alone (Fig. 5a). As shown in Fig. 5c, the half-life of degradation did not change significantly. These findings suggest that the potent inhibitory effect of Q39 on hypoxia-induced HIF-1α accumulation is not attributed to an increase in protein degradation.

Q39 Inhibits Hypoxia-Induced HIF-1α Protein Expression by Decreasing Its Synthesis, not by Promoting Its Degradation. a and b SMMC-7721 cells were exposed to hypoxia for 16 h followed by treatment with cycloheximide (CHX; 10 μg/mL) in the presence or absence of 5 μM Q39 for different periods. HIF-1α protein levels were determined by western blot analysis. c Quantitative densitometric analysis of results from (a) and (b). The dotted line represents the degradation half-life. d SMMC-7721 cells were treated with 20 μmol/L MG132 for 30 min and cultured in the presence of different concentrations of Q39 for 16 h under hypoxic (bottom) conditions. HIF-1α protein levels were determined by western blot. Representative of three independent experiments. e SMMC-7721 cells were pre-incubated with cycloheximide (CHX; 10 μg/mL) for 3 h in normal conditions, then placed in fresh medium and treated with or without 20 μM Q39 for different periods under hypoxic conditions. HIF-1α protein levels were analyzed by western blot
To examine whether Q39-mediated inhibition of HIF-1α accumulation was due to a reduction in protein synthesis, we introduced MG132, a specific proteasome inhibitor, to prevent ubiquitin-dependent HIF-1α degradation. Unexpectedly, we did not detect the ubiquitinated fraction of HIF-1α in the presence of MG132. However, MG132 treatment further induced HIF-1α protein accumulation under hypoxic conditions while the inhibitory effects of Q39 on hypoxia-induced HIF-1α protein levels were not abolished in the presence of MG132 (Fig. 5d), implicating an interference with the protein synthesis process. Thus, to analyze the rate of HIF-1α synthesis, SMMC-7721 cells were pre-treated with cycloheximide for 3 h under normoxia to block protein synthesis, then placed in fresh medium in the presence or absence of Q39 (20 μM) at different time points under hypoxic conditions, followed by immunoblotting of HIF-1α. As shown in Fig. 5e, Q39 treatment greatly decreased HIF-1α accumulation under hypoxia. This result clearly indicates that Q39 decreases the protein synthesis rate of HIF-1α. Together with the previous data, the results mentioned above suggest that Q39 inhibits hypoxia-induced HIF-1α protein expression by decreasing its synthesis but not by promoting its degradation.
Inhibition of HIF-1α by Q39 correlates with repression of the Akt/mTOR/4E-BP1 pathway
Previous studies have implicated the PI3K/Akt/mTOR pathway in the regulation of HIF-1α expression at the translational level [14–17]. In addition, our results show that pre-treatment of cells with LY294002, a PI3K inhibitor, dramatically suppressed hypoxia-induced HIF-1α protein accumulation (Fig. 6a). We were thus encouraged to explore whether the Akt signaling pathway was correlated with the inhibition of HIF-1α by Q39 treatment. To address the potential involvement of this pathway in Q39-mediated inhibition of HIF-1α accumulation, we measured the phosphorylation status of mTOR, its effectors p70S6K and 4E-BP1, and Akt. Treatment of SMMC-7721 cells with Q39 under hypoxia resulted in a dose-dependent dephosphorylation of Akt (Ser473), which correlated with the inhibition of phosphorylation of mTOR and 4E-BP1 (Fig. 6b, c). Moreover, Q39 did not affect the total protein levels of the analyzed corresponding phosphoproteins (Fig. 6b, c), indicating that this effect was specific to protein phosphorylation. It should be noticed that phosphorylation of p70S6K was not attenuated significantly after Q39 treatment. Collectively, these results suggest that 4E-BP1 plays a critical role in Q39-induced inhibition of HIF-1α without affecting the protein synthesis rate. Subsequently, we were encouraged to investigate the biological function of Q39-induced dephosphorylation of 4E-BP1.

AKT/mTOR/4E-BP1 signaling pathways contribute to hypoxia-induced HIF-1α expression. (a) SMMC-7721 cells were pretreated with various concentrations of LY294002 for 1 h in normal culturing conditions followed by exposure to hypoxia (3% O2) for 16 h. HIF-1α protein levels were analyzed by western blot. (b) SMMC-7721 cells were pretreated with different concentrations of Q39 (0–20 μM) or LY294002 for 1 h in normal conditions followed by exposure to hypoxia (3% O2) for 3 h. Phosphorylated Akt (Ser473) levels were analyzed by western blot. (c) mTOR/4E-BP1 signaling pathway involved in Q39-induced inhibition of HIF-1α. Cells were treated with 20.0 µM Q39 for 1 h in normal conditions followed by exposure to hypoxia (3% O2) for 3 h. Whole-cell lysates were collected and immunoblotted with primary antibodies. (d) SMMC-7721 cells were treated with 20.0 µM Q39. eIF4E was immunoprecipitated (IP) from cell lysates with anti-eIF4E antibody, and the resulting immunocomplexes were subjected to immunoblot analysis using the indicated antibodies
Hyperphosphorylation of 4E-BP1 disrupts the interaction between eIF4E translation initiation factor and translation repressor protein (4E-BP1), which results in the activation of cap-dependent translation. Therefore, we hypothesized that the dephosphorylation of 4E-BP1 caused by Q39 probably increased the binding of 4E-BP1 to eIF4E. As shown in Fig. 6c, no changes in expression of 4E-BP1 or eIF4E were observed in SMMC-7721 cells treated with Q39. Moreover, immunoprecipitation analysis revealed that the interaction between 4E-BP1 and eIF4E was enhanced after exposed to 20.0 µM Q39 (Fig. 6d). Taken together, these findings suggest that the inhibition of HIF-1α by Q39 correlates with the repression of the Akt/mTOR/4E-BP1 pathway.
Discussion
It has been well documented that aggressive solid tumors suffer from intratumoral hypoxia as a result of abnormalities in the microvasculature and a rapid expansion of tumor mass [18]. Hypoxia promotes tumor angiogenesis, and this process is predominantly accomplished by HIF-1α-induced overexpression of VEGF [19, 20]. As a result, a number of pharmacological strategies to inhibit hypoxia/HIF-1/VEGF activation have been developed, such as inhibiting the PI3K/Akt and MAPK pathways [21] or promoting proteasome-mediated degradation [22, 23]. Notably, the 1,2,4 benzotriazine 1,4-di-N-oxides, including the bioreductive drug tirapazamine and TX-402, which are Q39 analogues, have been identified as antiangiogenic compounds in vitro and are currently being investigated in vivo. The underlying mechanisms seemed to be associated with inhibition of VEGF production through the HIF-1 signaling pathway [24]. In the present study, we explored the anti-tumor activity of Q39 against hepatoma in vivo as well as the mechanism of Q39-mediated reduction of HIF-1α/VEGF expression in HCC cells. Importantly, the mechanisms by which Q39 inhibited hypoxia-induced HIF-1α protein expression seemed to involve blockage of HIF-1α protein accumulation via Akt/mTOR/4E-BP1 signaling pathways, but not the increase of HIF-1α protein degradation, therefore, suggesting a novel protein expression pathway and mode of regulation of HIF-1α at the translational level.
Hepatocellular carcinoma (HCC) is characterized as a highly chemoresistant cancer with no effective systemic therapy. Fortunately, our data show that Q39, as a single agent, potently suppresses tumor growth in an SMMC-7721 xenograft tumor model. The activity of Q39 contradicts that of bioreductive drugs, which have limited effect on tumor growth without combination methods to increase tumor hypoxia [25]. Thus, additional studies are required to identify the different mechanisms between Q39 and other hypoxic selective cytotoxins. HCC is a highly-vascularized tumor with rapid growth and frequent vascular invasion. Angiogenesis provides a target for novel prognostic and therapeutic approaches to HCC. VEGF seems to be the most important angiogenic factor in HCC known so far. Yoshiji et al. [26] have demonstrated in a study of HCC xenografts using nude mice that tumor growth in HCC was tightly regulated by VEGF expression. Additionally, several studies have further demonstrated overexpression of VEGF in HCC and its relationship with angiogenic activity or tumor progression in HCC [27, 28]. Most recently, Lu et al. reported that VEGF expression could be impaired via HIF-1α in HCC cells [29]. We have shown that Q39 significantly inhibited the growth of SMMC-7721 cells in vivo (Fig. 2c), accompanied with the decrease of HIF-1α and VEGF expression (Fig. 3g), as well as exhibited potential antiangiogenic activity (Fig. S1). Q39 is superior to other hypoxic selective cytotoxins (such as TPZ), making it a promising anticancer drug candidate demanding further investigation.
The expression of HIF-1α is tightly regulated through both protein degradation and protein synthesis. Our results indicate that Q39-treatment induced HIF-1α downregulation in SMMC-7721 cells without affecting the process of HIF-1α degradation. Q39 did not affect HIF-1α mRNA levels (Fig. 3c) nor decrease the phosphorylation status of p70S6K, which is recognized as regulating ribosomal biogenesis (Fig. 6c). Q39 was found to attenuate the expression of the phosphorylated 4E-BP1 (Fig. 6c). As a result, the interaction between eIF4E and 4E-BP1 was enhanced, as shown by the co-immunoprecipitation experiments (Fig. 6d). Collectively, the data mentioned above reveal a novel mechanism of Q39-induced reduction of HIF-1α accumulation at the translational level. Several lines of evidence suggest that the phosphorylation and activation of eIF4E, a key translation factor that mediates the initiation of mRNA translation, are involved in HIF-1α synthesis [17, 30]. It is reported that 4E-BP1 binds to eIF4E and inhibits eIF4E function. However, hyperphosphorylation of 4E-BP1 disrupts this binding, releasing eIF4E to associate with eIF4G and initiating cap-dependent translation [17]. Our data support the possibility that Q39 promotes the interaction between eIF4E and 4E-BP1 (Fig. 6d). Collectively, these results imply that the dysfunction of eIF4E caused by Q39-driven dephosphorylation of 4E-BP1 accounts for the inhibition of HIF-1α expression to some extent.
In summary, for the first time, we investigated the anti-tumor activity of Q39 against hepatoma in vivo and the mechanisms by which Q39 inhibits hypoxia-induced HIF-1α protein expression. These observations greatly extend our previous study by elucidating the molecular mechanisms of Q39, which will be significant to further understand this novel regulation of HIF-1α expression at the translational level.
Conclusion
We have shown for the first time that Q39 suppresses hypoxia-induced HIF-1α protein accumulation and VEGF expression in human hepatoma (SMMC-7721) cells but has no apparent inhibitory effects on HIF-1α mRNA levels. The mechanisms by which Q39 inhibits hypoxia-induced HIF-1α protein expression seem to be correlated with the blockage of HIF-1α protein accumulation via Akt/mTOR/4E-BP1 signaling pathways, but not the increase of HIF-1α protein degradation.
Abbreviations
- HIF-1:
-
hypoxia inducible factor-1
- VEGF:
-
vascular endothelial growth factor
- Akt:
-
oncogene from AKR mouse thymoma
- 4E-BP1:
-
initiation factor 4E-binding protein 1
- mTOR:
-
mammalian target of rapamycin
- p70S6K:
-
p70 S6 kinase
- PI3K:
-
phosphoinositide 3-kinase
- eIF4E:
-
eukaryotic translation initiation factor 4E
References
Sheng R, Xu Y, Weng QJ, Xia Q, He QJ, Yang B, Hu YZ (2007) Synthesis and cytotoxic activity of 3-phenyl-2-thio-quinoxaline 1, 4-dioxide derivatives in hypoxia and in normoxia. Drug Discov Ther 1:119–123
Weng Q, Wang D, Guo P, Fang L, Hu Y, He Q, Yang B (2008) Q39, a novel synthetic Quinoxaline 1, 4-Di-N-oxide compound with anti-cancer activity in hypoxia. Eur J Pharmacol 581:262–269
Semenza GL (2002) HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med 8:S62–S67
Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9:677–684
Fang J, Ding M, Yang L, Liu LZ, Jiang BH (2007) PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal 19:2487–2497
Jackson MW, Roberts JS, Heckford SE, Ricciardelli C, Stahl J, Choong C, Horsfall DJ, Tilley WD (2002) A potential autocrine role for vascular endothelial growth factor in prostate cancer. Cancer Res 62:854–859
Soker S, Kaefer M, Johnson M, Klagsbrun M, Atala A, Freeman MR (2001) Vascular endothelial growth factor-mediated autocrine stimulation of prostate tumor cells coincides with progression to a malignant phenotype. Am J Pathol 159:651–659
Li XM, Tang ZY, Zhou G, Lui YK, Ye SL (1998) Significance of vascular endothelial growth factor mRNA expression in invasion and metastasis of hepatocellular carcinoma. J Exp Clin Cancer Res 17:13–17
Li ZD, Liu LZ, Shi X, Fang J, Jiang BH (2007) Benzo[a]pyrene-3, 6-dione inhibited VEGF expression through inducing HIF-1alpha degradation. Biochem Biophys Res Commun 357:517–523
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107:43–54
Beibei Fu, Xue J, Li Z, Shi X, Jiang B-H, Fang J (2007) Chrysin inhibits expression of hypoxia-inducible factor-1α through reducing hypoxia-inducible factor-1α stability and inhibiting its protein synthesis. Mol Cancer Ther 6:220–226
Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL (2000) Modulation of hypoxia-inducible factor 1 α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60:1541–1545
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL (2001) HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1α (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 21:3995–4004
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT (2002) Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol Cell Biol 22:7004–7014
Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL (2006) Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 12:122–127
Topisirovic I, Ruiz-Gutierrez M, Borden KLB (2004) Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res 64:8639–8642
Belozerov VE, Van Meir EG (2005) Hypoxia inducible factor-1: a novel target for cancer therapy. Anticancer Drugs 16:901–909
Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang B-H (2005) Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J 19:342–353
Powis G, Kirkpatrick L (2004) Hypoxia inducible factor 1α as a cancer drug target. Mol Cancer Ther 3:647–654
Dai M, Miao ZH, Ren X, Tong LJ, Yang N, Li T, Lin LP, Shen YM, Ding J (2009) MFTZ-1 reduces constitutive and inducible HIF-1alpha accumulation and VEGF secretion independent of its topoisomerase II inhibition. J Cell Mol Med, in press.
Li MH, Miao ZH, Tan WF, Yue JM, Zhang C, Lin LP, Zhang XW, Ding J (2004) Pseudolaric acid B inhibits angiogenesis and reduces hypoxia-inducible factor 1alpha by promoting proteasome-mediated degradation. Clin Cancer Res 10:8266–8274
Hagen T, D’Amico G, Quintero M, Palacios-Callender M, Hollis V, Lam F, Moncada S (2004) Inhibition of mitochondrial respiration by the anticancer agent 2-methoxyestradiol. Biochem Biophys Res Commun 322:923–929
Nagasawa H, Mikamo N, Nakajima Y, Matsumoto H, Uto Y, Hori H (2003) Antiangiogenic hypoxic cytotoxin TX-402 inhibits hypoxia-inducible factor 1 signaling pathway. Anticancer Res 23:4427–4434
McKeown SR, Cowen RL, Williams KJ (2007) Bioreductive drugs: from concept to clinic. Clin Oncol 19:427–442
Yoshiji H, Kuriyama S, Yoshii J, Yamazaki M, Kikukawa M, Tsujinoue H, Nakatani T, Fukui H (1998) Vascular endothelial growth factor tightly regulates in vitro development of murine hepatocellular carcinoma cells. Hepatology 28:1489–1496
Moon WS, Rhyu KH, Kang MJ, Lee DG, Yu HC, Yeum JH, Koh GY, Tarnawski AS (2003) Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma? Mod Pathol 16:552–557
Poon RT, Lau CP, Cheung ST, Yu WC, Fan ST (2003) Quantitative correlation of serum levels and tumor expression of vascular endothelial growth factor in patients with hepatocellularcarcinoma. Cancer Res 63:3121–3126
Lu L, Yang Z, Zhu B, Fang S, Yang X, Cai W, Li C, Ma JX, Gao G (2007) Kallikrein-binding protein suppresses growth of hepatocellular carcinoma by anti-angiogenic activity. Cancer Lett 257:97–106
Jin X, Jin HR, Lee D, Lee JH, Kim SK, Lee JJ (2008) A quassinoid 6alpha-tigloyloxychaparrinone inhibits hypoxia-inducible factor-1 pathway by inhibition of eukaryotic translation initiation factor 4E phosphorylation. Eur J Pharmacol 592:41–47
Acknowledgments
This work was supported by The National Natural Science Foundation (No. 30873096 and 20602030), Zhejiang Provincial Natural Science Foundation (No. R2080326 and Z2090053), and Education Department Foundation of Zhejiang Province (No. 20070166 and No. 20080116). We also extend our heartfelt thanks to Dr. Huijun Zhou and Jiali Zhang for their help.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Qinjie Weng and Jun Zhang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1
Q39 exhibits potential antiangiogenic activity. Inhibition of angiogenesis was determined using the CAM assay. The sterile filter paper square saturated with Q39 (2 and 8 μg), TPT (as a positive control) or physiological saline solution was placed in areas between vessels but never onto any large vessels. After 48 h, the CAMs were carefully isolated and were fixed in methanol/acetone. Results of the samples were photographed (PDF 29 kb)
Rights and permissions
About this article
Cite this article
Weng, Q., Zhang, J., Cao, J. et al. Q39, a quinoxaline 1,4-Di-N-oxide derivative, inhibits hypoxia-inducible factor-1α expression and the Akt/mTOR/4E-BP1 signaling pathway in human hepatoma cells. Invest New Drugs 29, 1177–1187 (2011). https://doi.org/10.1007/s10637-010-9462-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-010-9462-y