
Summary
Hepatocellular carcinoma (HCC) remains a lethal treatment-resistant cancer with a median survival of <6 months in patients not considered candidates for radical surgical treatments. SB-715992 is a novel cytotoxic agent implicated in the inhibition of mitotic kinesin spindle protein (KSP). Based on evidence from preclinical models and phase I trials, we conducted a phase II trial of SB-715992 in chemo-naïve patients with advanced HCC. A non-randomized, non-blinded multicentre two-stage phase II study was completed examining the efficacy, toxicity, and pharmacokinetics of SB-715992 at 18 mg/m2 IV q 3 weeks, in patients with measurable locally advanced, metastatic or recurrent HCC. The predictive value of KSP in archival tumour was assessed. Fifteen patients with metastatic HCC received a median of 3 cycles of SB-715992. The most common grade 3+ toxicities were granulocytopenia, leukocytopenia, diarrhea and liver transaminase rise. Overall confirmed response rate was 0%. Seven (46%) patients had a best response of stable disease at the 8-week evaluation (median duration 3.9 months) Median time to progression was 1.61 months (95%CI = 1.31–3.94 months) SB-715992 plasma concentrations were comparable to those observed in the phase I studies. Expression of KSP by immunohistochemistry was observed in only four of eight evaluable samples with strong expression reported in only two. No correlation was observed between intensity of KSP staining and clinical outcome. Among these patients with preserved hepatic function and good performance status, SB-715992 was generally well tolerated. However, no conclusive evidence of benefit was seen with SB-715992 monotherapy in HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) remains a leading cancer worldwide and its incidence is rising, with the highest rates observed in Asia and Africa secondary to endemic hepatitis B and C carrier rates [1]. Over the past two decades, a marked upward trend of incidence in North America has also been observed [2]. Unfortunately, HCC is typically diagnosed late in the course of patients with chronic liver disease, with an overall median survival following diagnosis of <1 year [3]. The prognosis of HCC is poor due to both tumour factors and the underlying diseased liver that often predisposes a patient to HCC. Hepatic resection is the treatment of choice in selected cases, with transplantation being an alternative curative approach in others (3-year survivals of approximately 70%) [4]. However, long term prognosis remains unsatisfactory because of the high incidence of recurrence even after curative resection with 5-year actuarial recurrence rate of 75–100% [5]. These recurrence rates are thought to be due to re-growth of residual microscopic disease and/or the development of new primaries in the diseased liver. Patients with liver-limited disease not eligible for resection or transplant may benefit from other local approaches such as ethanol injection, radiofrequency ablation or chemo-embolization. These are widely used techniques that lead to objective responses in tumours but their impact on overall survival remains controversial [6, 7].
A median survival of 6 months is estimated in patients with HCC who are not considered candidates for radical treatments, but this can vary depending of prognostic features [8–10, 12]. Advanced HCC is refractory to standard forms of chemotherapy. This is largely attributed to inherent drug resistance and limitations due to underlying hepatic dysfunction. Although doxorubicin is commonly used as a first-line systemic treatment option for advanced disease, overall response rates and duration of response remain poor. As a single agent, doxorubicin is associated with a response rate of 15% and a very modest improvement in survival when compared to best supportive care [8]. With combination therapies, response rates improve to 20% to 35%; however, there is no proven survival benefit [9, 10]. Just recently, Llovet et al. reported a survival benefit for HCC patients treated with Sorafenib, the multitargeted tyrosine kinase inhibitor, vs placebo in a large European-based phase III trial, consisting of patients with advanced HCC [11]. Overall survival for these patients with preserved liver function was 10.7 months on the sorafenib arm vs 7.9 months with placebo (p = 0.00058). It is expected that this trail could lead to a new standard of care in select HCC patients.
The study reported here was initiated before the sorafenib results were known. Clearly, there was and still is a need for novel agents, which will have improved activity in unresectable locally advanced or metastatic HCC. It would follow that the best candidates for entry into studies aimed at investigating promising new therapies should be patients who do not fulfill the stringent criteria for benefit of radical therapies but excluding those at the terminal stages of their disease. This “intermediate group” of patients constitutes the majority of patients diagnosed with HCC today [12].
SB-715992 (ispinesib, Cytokinetics Inc.) is a cytotoxic compound with a novel mechanism of action, namely, inhibition of the mitotic kinesin spindle protein (KSP), a member of the kinesin superfamily which is necessary in the initial stages of mitotic spindle formation [13]. KSP is necessary in the initial stages of mitosis for the separation of spindle poles and eventual formation of distinct daughter cells. KSP is overexpressed in proliferating cells relative to non-proliferating cells and tumour tissue relative to non-proliferating cells and normal tissue respectively [14]. Inhibition by SB715992 involves an enzyme–inhibitor interaction that results in a decrease in the rate of release of adenosine disphosphate (ADP) from KSP with consequent inhibition of monopolar spindle formation and induction of apoptosis [15]. Phase I studies of SB715992 in patients with solid tumours have been completed [16, 17]. SB715992 is well tolerated when administered on a once every 21-day schedule. Neutropenia was dose-limiting and maximum tolerated dose was reached at the 18 mg/m2 q 21 days [16]. Other adverse events include grade 1 diarrhea, grades 1 and 2 fatigue and grade 1 injection site reactions. No significant gastrointestinal or neurotoxicity has been demonstrated. A sustained minor response was observed in one patient with hepatocellular carcinoma.
We report here on the results of a non-randomized, non-blinded multicentre phase II study examining the efficacy, toxicity, and pharmacokinetics (PK) of SB-715992 in patients with measurable locally advanced, metastatic or recurrent hepatocellular carcinoma for whom no curative therapy was available. The predictive value of KSP in archival tumour as well as the effects SB715992 has on the cytoskeleton of circulating peripheral blood mononuclear cells were assessed.
Patients and methods
These investigations were performed after approval of local institutional research ethics boards as well as the NCIC-CTG ethics board. Eligible patients were at least 18 years of age, able to give informed written consent and with a life expectancy of at least 12 weeks. They had histologically documented hepatocellular carcinoma with locally advanced or recurrent disease with archival paraffin tumour specimen available for study. Patients had a hepatic reserve of Child–Turcotte–Pugh Class A or better, an ECOG performance status of 0, 1 or 2, absolute granulocytes of ≥1.5 × 109/l, platelets ≥80 × 109/l, aspartate aminotransaminase less than five times the upper limit of normal (ULN), creatinine clearance ≥50 ml/min and bilirubin less than or equal to two times the ULN. Prior intra-hepatic chemotherapy was permitted but prior systemic chemotherapy was not. Patients were ≥4 weeks from any major surgery, radiation therapy, local ablative therapy or intra-hepatic chemotherapy. They were required to have unidimensionally measurable disease and be on no other concurrent anti-cancer treatment.
Study evaluations and treatments
History and physical exam, complete blood count (CBC) and differential, INR, serum biochemistry, alpha fetoprotein (AFP), chest radiograph, triphasic abdominal CT scan or magnetic resonance imaging, and any other imaging necessary to document disease was done at baseline. Physical exam, including vital signs, weight and performance status, international normalized ratio AFP and biochemistry were repeated every 3 weeks while CBC was collected weekly. Pharmacokinetic blood samples were collected cycle 1 only in all patients. Peripheral blood mononuclear cells (PBMC) were collected for molecular correlative studies in cycle 1 in consenting patients. Tumour response was assessed using Response Evaluation Criteria In Solid Tumours (RECIST) criteria [18] every 6 weeks. Adverse events were graded using the NCI Common Terminology Criteria for Adverse Events Version 3.0 (CTCAE).
Drug administration
SB-715992 (ispinesib) was administered as a 1-h intravenous infusion in a dose of 18 mg/m2 once every 3 weeks on an out-patient basis and was not escalated. Doses were reduced for hematologic and other toxicities. Patients received ongoing supportive and palliative care (e.g. nutritional support, pain control) as indicated throughout the study. Colony stimulating factors (CSFs) were not prohibited but their use could not substitute for a required dose reduction.
Statistical design
This was a two-stage phase II study to investigate the efficacy of SB-715992 in previously untreated patients with metastatic or recurrent malignant hepatocellular carcinoma. Secondary objectives were to determine the adverse events and tolerability of SB-715992 when given to patients with advanced hepatocellular carcinoma and to describe time to disease progression and overall patient survival.
A multinomial stopping rule incorporating both response and early progression (defined as progression at or before 6 weeks of therapy) was employed in a 2-stage design. We used the response hypotheses of Ho ≤ 5% and Ha ≥ 20% and progression hypotheses of Ho ≥ 60% and Ha ≤ 40%. After entering 15 evaluable patients the drug would be rejected at the end of the first stage of accrual if no responses and 10 or more early progressors were seen. If the above criteria were not met in the first stage of accrual, 15 additional response evaluable patients, for a total of 30 patients, would be accrued. At the end of the second stage, we would accept the drug as active in this schedule if one of the following was observed: ≥4 responses or ≤13 early progressions in the 30 evaluable patients.
Pharmacokinetics
Pharmacokinetic studies were included to examine PK parameters in a patient population with impaired liver function. If the activity of SB-715992 proved to be promising, scatterplots and means, standard deviations, and confidence intervals would be constructed to explore differences between responders and non-responders, and patients who did or did not experience unacceptable toxicity. Serial blood samples (3 ml/sample) were collected during cycle 1 day 1: pre-dose, immediately following completion of the infusion, post infusion (30–60 min, 1.5–2.5 h, 4–6 h, 20–29 h, and 36–48 h). Cycle 2 PK samples were only collected if the dose of SB-715992 was adjusted for any reason following cycle 1.
The method for the determination of SB715992 (ispinesib) in human plasma has been validated over the range 0.1 to 100 ng/ml using HPLC-MS/MS. SB715992 was extracted from 50 μl of human plasma by protein precipitation using 75/25 acetonitrile/10 mM ammonium formate (pH 3) containing an isotopically labeled internal standard ([2H4]-ispinesib). Extracts were analyzed by HPLC-MS/MS using a TurboIonSpray™ interface and multiple reaction monitoring.
A population pharmacokinetic analysis was conducted using NONMEM (Globomax LLC; Hanover, MD) on Phase I SB715992 data following an 18-mg/m2 dose, the maximum tolerated dose from a once (1-h intravenous infusion) every 21-day schedule. Once a final model was selected, it was validated using the posterior predictive check (PPC) method. Upon validation of the final population model, it was used to simulate plasma SB715992 concentration–time profiles resulting from a dose of 18 mg/m2. A post-hoc median profile with 95% confidence interval were simulated using the population pharmacokinetic model developed using Phase I data. Observed study data were overlaid on the simulated profile to determine if the observed data exhibit the central tendency and variability of the Phase I data obtained at the same dose level.
Pharmacodynamics
Archival tumour specimens
For evaluation of tumoural hEg5 (Kinesin-related motor protein Eg5 or kinesin-line spindle protein [KSP]), formalin-fixed, paraffin-embedded tissue were cut in 4-μm sections and placed onto positively charged slides. Slides were deparaffinized in xylene, and rehydrated in decreasing concentrations of ethanol/water. Following antigen retrieval and blocking, sections were incubated for 30 min with an anti-hEg5 monoclonal antibody (kindly provided by Dr. Yun-Fu Hu, GlaxoSmithKline) at a dilution of 1:5,000. Immunohistochemical staining was performed using an Envision+polymer (rabbit) and the Dako Autostainer Universal Staining System. Slides were counterstained with 50% Mayer’s hematoxylin. Human tonsillar tissue was used as positive control for hEg5 staining. The hEg5 stained slides were read as positive or negative. Staining intensity was scored by two independent reviewers in a semi-quantitative manner. The positive cases were divided in weak staining (1+) and strong staining (2+).
Cytoskeletal changes in PBMCs by immunohistochemistry
For analysis of cytoskeletal morphologic changes following drug treatment, peripheral blood mononuclear cells (PBMC) were isolated from whole blood at the following times around dose 1 of cycle 1; pre-treatment, immediately prior to the end of the infusion, and then 1, 4, and 24 h after the end of the infusion. At each of the indicated times, 8 ml of blood was drawn in CPT® tubes (Becton Dickinson cat. # 362753) for isolation of PBMC. Cytospins were made and fixed at each of the treating institutions before being shipped to the central laboratory for analysis.
Analysis consisted of simultaneous fluorescent immunohistochemical assessment of beta-tubulin, phosphohistone H3, and chromatin. Briefly, PBMC’s were incubated with primary antibodies (e.g. beta-tubulin, and phosphohistone H3) for 24 h at 4°C. After thorough washing, slides were incubated with fluorescent-labeled secondary antibodies for 3 h at room temperature. After another round of washing, slides were incubated with DAPI (Sigma D9542) for labeling of chromatin, followed by a final round of washing. Finally, the partially dried slides were covered with glass cover slips using Hard Set VectaShield (Vector Laboratories, Burlingame, CA) mounting media.
Stained slides were imaged using an Olympus AX70 automated upright fluorescent microscope fitted with a Q imaging Retiga EXi CCD camera, and image analysis was performed using Image Pro Plus software (Media Cybernetics, Inc., Silver Spring, MD). Separate images were acquired for the specific target protein (e.g. tubulin, and phosphohistone H3) and DNA, and the final image was obtained by merging the separate data files. With each set of slides to be analyzed, human cultured cell line controls (Hela cells), both positive (i.e. with 1° antibody) and negative (i.e. without 1° antibody), were included to assess the adequacy and specificity of the staining procedure.
Results
Patient characteristics
A total of 15 patients were enrolled in this clinical trial over an 11-month period (Table 1). Fourteen (93%) of the patients were men and the median age was 57 years (range 22–73). Five patients (33%) were non-Hepatitis B and C. Most patients had an ECOG performance score of 0 or 1 (80%). All patients had extrahepatic metastases. The criteria for the stopping rule were met after the first stage and the study was permanently closed to accrual.
Outcome measures
At the end of stage 1, 15 patients were evaluable for response. All 15 patients discontinued protocol therapy due to progressive disease. The overall confirmed response rate was 0% [95% confidence interval (CI) 0% to 18.1%]. There were two patients who appeared to derive clinical benefit with durable stable disease. Both had bulky progressing multifocal HCC at the time of study entry and tolerated drug well until discontinued with progressive disease at 14 and 10 months. One of these stable patients had an elevated AFP that increased slowly over treatment period twofold. The second did not produce AFP as a tumor marker. Overall, only 7 (46%) patients had a best response of stable disease at the 8-week evaluation [median duration 3.9 months (range 2.6–9.5)]. Eight patients had early progressive disease. Based upon pre-defined stopping rules, 10 early progressors were required for discontinuing study at the end of stage 1. However, since two additional patients with borderline stable disease criteria at the 8-week evaluation were observed to have clear progression soon afterwards, it was determined that we had met our stopping rules and the data did not justify continuing to stage 2.
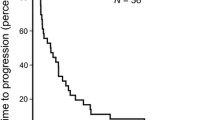
The median survival of patients on study was 11.6 months and the median time to progression was 1.61 months (95%CI = 1.31–3.94 months; Fig. 1).

a Kaplan–Meier distribution of time to death (n = 15); b Kaplan–Meier distribution of time to disease progression (n = 15)
Dose intensity
A median of three cycles of therapy were given (range 1–14) and 60% of patients received ≥90% of the 6.0 mg/m2/week planned dose intensity. Seven patients received four or more cycles and only five patients (33%) were treated beyond cycle 6. Doses were reduced in six patients (40%) due to grade 3 events [elevation of creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT), infection, rash and diarrhea].
Adverse events
Adverse events were evaluable in all 15 patients (Table 2). The most common hematologic grade 3+ toxicities were granulocytopenia (87%) and leukocytopenia (53%). Eleven patients (73%) experienced grade 1 or 2 fatigue and 9 patients (60%) experienced grade 1 or 2 injection site reaction. Grade 3+ transaminitis occurred in 6 patients (40%). No grade 5 adverse events were observed. The toxicity profile was similar to that previously observed in the phase I setting.
Correlative studies
Pharmacokinetics
Using a population pharmacokinetic model derived from the phase I data of the maximum tolerated dose of 18 mg/m2 every 21 days, a comparison of the sparsely sampled observed data in this study was made (n = 8)). In this cohort of patients with underlying compensated liver disease, SB-715992 plasma concentrations following an 18-mg/m2 dose were comparable to those observed in the phase I studies, as shown in Fig. 2. The fixed and random effects of the population pharmacokinetic model adequately describe the central tendency and variability of the observed data.

Pharmacokinetics: SB-715992 observed concentrations at 18 mg/m2
Immunohistochemistry
Eight archival tumour specimens were suitable for immunohistochemical analysis of KSP staining by tumoural hgE5 expression (Fig. 3). Summary of staining results vs clinical outcome are shown in Table 3. By maximal staining intensity observed, strong staining (2+) was identified in two specimens (25%), weak staining (1+) in two specimens (25%) and negative staining observed in the remaining four specimens (50%).

Eight patient archival tumour specimens showing immunohistochemical analysis of tumoural hgE5 expression
Pharmacodynamics
Serial PBMC samples were available from four patients for analysis of cytoskeletal morphologic changes following treatment with SB715992. Figure 4 depicts the characteristic morphologic changes seen in cultured Hela cells grown in the presence of SB-715992. As shown in the figure, dividing cells treated with the drug display catastrophic mitoses or monasters, while non-dividing cells show no obvious phenotype. No monasters were seen in any of the PBMC samples collected from the four subjects.

Morphologic cytoskeletal changes seen in cultured Hela cells grown in the presence of SB-715992. (a) untreated Hela cells; (b) 10 nM SB-715992 × 16 hours
Discussion
Hepatocellular carcinoma remains a lethal treatment-resistant cancer with a median survival of <6 months in patients not considered candidates for radical surgical treatments. SB-715992 is a novel cytotoxic agent implicated in the inhibition of mitotic kinesin spindle protein. Based upon evidence from encouraging preclinical models and phase I clinical experience, we conducted a phase II trial of SB-715992 in chemo-naïve patients with advanced hepatocellular carcinoma.
In this study, no conclusive evidence of benefit was seen with SB-715992 monotherapy. All patients on trial had metastatic disease. While there was no evidence of objective response benefit, non-progression was observed in seven patients (47%) for a median duration of 3.9 months. Among these patients with preserved hepatic function and good performance status, SB-715992 was generally well tolerated. Notable grade 3/4 adverse events included granulocytopenia, transaminitis and diarrhea. Grade 1 or 2 fatigue was common. No grade 5 events were observed and no patients came off study for treatment toxicity. Plasma concentrations of SB715992 achieved in this study were comparable to those observed in the Phase I study that determined the maximum tolerated dose to be 18 mg/m2 every 21 days [16].
We attempted to evaluate cytoskeletal morphologic changes in PBMCs by immunohistochemistry. The fluorescent immunohistochemistry was negative in all four evaluable samples. This finding has since been observed across seven clinical trials of SB-715992. Because circulating PBMC’s are mature cells, and since SB715992 only affects actively dividing cells, it is not surprising that no drug-induced morphologic changes were observed in our patients. To confirm that mitotic cell division is required for SB-715992 induced monaster formation, donor lymphocytes were mitogen stimulated with phytohaemagglutinin (PHA) in the presence and absence of SB-715992 and monopolar spindles were observed only in the PHA-stimulated cells treated with drug (Synold, personal communication; data not shown).
One may consider potential explanations for why SB-715992 failed to demonstrate benefit in this patient population. HCCs are inherently resistant tumours to cytotoxic chemotherapy [19] and are known to express the multidrug-resistant gene MDR-1 [19, 20]. Preclinical studies have suggested that SB-715992 may be a substrate for MDR-1 resulting in a variation in response [21]. In addition, the cytotoxicity of SB-715992 is mediated through inhibition of KSP and this agent is highly selective for KSP relative to other members of the kinesin superfamily [14] However, in our cohort, expression of KSP by immunohistochemistry was observed in only four of eight evaluable samples with strong expression reported in only two samples. Furthermore, no clear correlation was observed between intensity of KSP staining and clinical outcome (Table 3).
Finally, the median survival in our cohort of 15 patients at 11.6 months compares favorably with the Sorafenib arm of the recently reported trial by Llovet et al. (10.7 months [11]), and while the studies had similar eligibility criteria, the small numbers in our study make comparisons quite problematic. There may also be prognostic differences between the North American HCC population studied here compared with the European-based trial. And while we checked for radiological progression more frequently than Llovet et al., the comparisons of the median time to progression is quite marked, also supporting our conclusion that SB-715992 is inactive in HCC. (Median TTP of 1.6 months in our study vs the phase III data of 5.5 months (sorafenib-arm) and 2.8 months (placebo-arm)).
The identification of appropriate and effective therapies in HCC represents a significant challenge. Important considerations in designing trials in HCC include patient selection and the use of appropriate end points. It is recognized that there may be a limited correlation between tumour response and necrosis and traditional RECIST methods of response assessment. In addition to a better understanding of the mechanisms of hepatocarcinogenesis, there is a need to develop more reliable methods of translating preclinical findings to predictors of response and benefit.
References
Parkin DM, Bray F, Ferlay J et al (2005) Global cancer statistics, 2002. CA Cancer J Clin 55(2):74–108
El-Serag HB, Mason AC (1999) Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med 340(10):745–750
Perry JF, Strasser SI et al (2003) Pharmacotherapy of hepatocellular carcinoma. Expert Opin Pharmacother 4(12):2175–2185
Bismuth H, Majno PE et al (1999) Liver transplantation for hepatocellular carcinoma. Semin Liver Dis 19(3):311–322
Poon RT, Fan ST et al (2000) Long-term prognosis after resection of hepatocellular carcinoma associated with hepatitis B-related cirrhosis. J Clin Oncol 18(5):1094–1101
Camma C, Schepis F et al (2002) Transarterial chemoembolization for unresectable hepatocellular carcinoma: metaanalysis of randomized controlled trials. Radiology 224(1):47–54
Llovet JM, Bruix J (2003) Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 37(2):429–442
Lai CL, Wu PC et al (1988) Doxorubicin versus no antitumor therapy in inoperable hepatocellular carcinoma. A prospective randomized trial. Cancer 62(3):479–483
Johnson PJ (2003) Are there indications for chemotherapy in hepatocellular carcinoma? Surg Oncol Clin N Am 12(1):127–134
Yeo W, Zee B, Leung WT et al (2004) A phase III study of doxorubicin (A) versus cisplatin (P)/ interferonA-2B (I)/ doxorubicin (A)/fluorouracil (F) combination chemotherapy (PIAF) for inoperable hepatocellular carcinoma (HCC). Proc Am Soc Clin Oncol 22:2004 (abstr 4026)
Llovet J, Ricci S, Mazzaferro V, Hilgard P, Raoul J, Zeuzem S, Poulin-Costello M, Moscovici M, Voliotis D, Bruix J, for the SHARP Investigators (2007) Sorafenib improves survival in advanced Hepatocellular Carcinoma (HCC): results of a Phase III randomized placebo-controlled trial. J Clin Oncol 25:abstr LBA1
Llovet JM, Bruix J (2000) Prospective validation of the Cancer of the Liver Italian Program (CLIP) score: a new prognostic system for patients with cirrhosis and hepatocellular carcinoma. Hepatology 32(3):679–680 (letter)
Wood KW, Cornwell WD, Jackson JR (2001) Past and future of the mitotic spindle as an oncology target. Curr Opin Pharmacol 1:370–377
Lee Y, Jia Z, Sakowicz R (2002) Inhibitors of the mitotic kinesin KSP: biochemical mechanism of action. Proc Am Assoc Cancer Res 43:A325
Vale RD, Milligan RA (2000) The way things move: looking under the hood of molecular motor proteins. Science 288:88–95
Chu QS, Holen KD, Rowinsky EK, Wilding G, Volkman JL, Orr JB, Williams DD, Hodge JP, Sabry J (2004) Phase I trial of novel kinesin spindle protein (KSP) inhibitor SB-715992 IV Q 21 days. Proc Am Soc Clin Oncol 22:A2078
Heath EI, Alouisi A, Eder JP, Valdivieso M, Vasist LS, Appleman L, Bhargava P, Colevas AD, Lorusso PM, Shapiro G (2006) A phase I dose escalation trial of ispinesib (SB-715992) administered days 1–3 of a 21-day cycle in patients with advanced solid tumors. Proc Am Soc Clin Oncol 24:A2026
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, Van Oostrom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors (RECIST guidelines). J Natl Cancer Inst 92:205–216
Huang M, Liu G (1999) The study of innate drug resistance of human hepatocellular carcinoma Bel7402 cell line. Cancer Lett 135:97–105
Kuo MT, Zhao JY, Teeter LD et al (1992) Activation of multidrug resistance (P-glycoprotein) mdr3/mdr1a gene during the development of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell Growth Differ 3:531–540
Johnson RK, McCabe FL, Caulder E et al (2002) SB-715992, a potent and selective inhibitor of the mitotic kinesin KSP, demonstrates broad-spectrum activity in advanced murine tumors and human tumor xenografts. Proc Am Assoc Cancer Res 43:A1335
Acknowledgement
The National Cancer Institute of Canada Clinical Trials Group supported the design, conduct, evaluation and manuscript preparation of this trial; drug was supplied by the National Cancer Institute Division of Cancer Therapy Evaluation Program (CTEP).
Author information
Authors and Affiliations
Corresponding author
Additional information
Knox and Sharlene are co-principal investigators.
Rights and permissions
About this article
Cite this article
Knox, J.J., Gill, S., Synold, T.W. et al. A phase II and pharmacokinetic study of SB-715992, in patients with metastatic hepatocellular carcinoma: a study of the National Cancer Institute of Canada Clinical Trials Group (NCIC CTG IND.168). Invest New Drugs 26, 265–272 (2008). https://doi.org/10.1007/s10637-007-9103-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-007-9103-2