
Summary
Fenretinide is a synthetic retinoid with chemotherapeutic activity against various malignancies. Upon oral administration to animals, fenretinide was found to be incompletely absorbed and excreted primarily in feces. The purpose of this study was to determine the possible reasons for poor oral absorption of fenretinide using Caco-2 cell monolayers. To achieve this purpose, a solid dispersion of fenretinide with Povidone K25 was used. The apparent permeability coefficient (P app) of fenretinide across Caco-2 monolayers in the presence of bovine serum albumin (BSA) in the receiver was determined. Apical to basolateral (AP-BL) and basolateral to apical (BL-AP) flux studies were performed to determine the role of an efflux mechanism. In the presence of 4% BSA in the receiver, the P app was found to be (8.8 ± 0.5) × 10−8 cm/sec. The AP-BL flux increased linearly with an increase in fenretinide concentration (125–640 μM) in the presence of 4% BSA in the receiver. Efflux and paracellular pathways played an insignificant role in the permeability of fenretinide. A significant amount of drug, approximately 13–15% of the initial amount accumulated in the cell membrane. The amount of fenretinide in the donor decreased by 16% over a 3 h period. However, only 0.12% of the initial amount was found in the receiver. Also, the P app increased with an increase in plasma protein concentration in the receiver. On the basis of these results, the poor permeability of fenretinide can be attributed to its accumulation in the lipophilic cell membrane and poor partitioning into the receiver medium.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fenretinide (M w = 391.6 Da) is a synthetic retinoid [N-(4-hydroxyphenyl)retinamide or 4-HPR], that has been shown to inhibit carcinogenesis in animal models for breast, bladder, lung, ovary and prostate cancers [1, 2]. Fenretinide (Fig. 1) is less toxic and teratogenic than other retinoids [3], thereby making it one of the most promising retinoid compounds for chemoprevention [4]. A phase III clinical trial suggested a reduced risk of contralateral and ipsilateral breast cancer in premenopausal women treated with fenretinide [5, 6]. The exact mechanism by which fenretinide elicits its cytotoxic action is still unclear. Recent studies have indicated that both receptor-dependent and –independent pathways might be involved in fenretinide induced chemopreventive and cytotoxic effects [7].
Fenretinide has been administered orally as an oil formulation in soft gelatin capsules during animal studies and clinical trials. Fenretinide in its current dosage form exhibits poor oral bioavailability and is incompletely absorbed after oral administration [8, 9]. In order to achieve chemotherapeutic levels, multi-gram quantities (as a very large capsule or as multiple capsules) of fenretinide are administered. The various factors that might account for poor absorption of fenretinide from the gut include poor solubility and/or dissolution, a significant paracellular component in passive diffusion, drug efflux by P-gp or similar proteins, absorption via a saturable carrier-mediated transporter, extensive metabolism, and accumulation in the cell membrane. If the mechanisms of fenretinide absorption are completely understood, various strategies can be employed to achieve higher fenretinide plasma concentrations. A majority of drugs are transported across the intestinal epithelium by passive diffusion and/or carrier-mediated transport. Passive transport may occur either through the cell after partitioning (transcellular route) or by diffusion through water filled pores in between the cells (paracellular route). The presence of tight junctions coupled with a limited surface area (0.01–0.1% of that of the transcellular route) results in a very low efficiency of the paracellular route in terms of absorption. In case of carrier-mediated transport, a limited availability of the carrier proteins might limit drug absorption. Also, certain transporters like PEPT1, MCT1, and OATP-B demonstrate pH dependence for transporting substrates [10].

Structure of fenretinide
The objective of this investigation was to carry out transport studies of fenretinide across Caco-2 cell monolayers with an aim to elucidate the reasons for its poor oral absorption.
Materials and methods
Materials
Fenretinide was purchased from Sigma Chemicals (St. Louis, MO). Caco-2 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD). Bovine serum albumin (BSA), retinol binding protein (RBP), all trans-retinol, and trypsin-EDTA solution were purchased from Sigma (St. Louis, MO). Antipyrine and HEPES buffer were from Spectrum (Gardena, CA). Dulbecco's modified Eagle medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, NY). Penicillin-Streptomycin and L-glutamine were obtained from Cellgro (Herndon, VA). Transwell®-col inserts (24 mm diameter, 0.4 μm pore size) and cluster plates were from Costar (Bedford, MA). HPLC grade solvents were purchased from Fisher (New Jersey). All materials were used as obtained without further purification.
Preparation of fenretinide solution
Fenretinide is practically insoluble in water. Therefore, a solid dispersion of fenretinide in Povidone K 25 (28–34 kDa) at a ratio of 1:20 with butylated hydroxytoluene (BHT) (0.005% w/w, in ethanol; as antioxidant) was used. The maximum concentration of fenretinide used in the transport study was 250 μg/mL (640 μM). Precipitation of fenretinide in the transport buffer was negligible over a 5 h period, if any.
Characterization of physicochemical properties
The n-octanol-water partition coefficient (log P) was determined by a shake flask method at 25°C according to a published procedure [11]. The pK a value for fenretinide was estimated to be 9.96 using the ACD/Solaris Software (Ontario, Canada). Solubility studies of fenretinide and fenretinide-Povidone K 25 solid dispersion system were carried out by the shake flask method in Hanks Balanced Saline Solution (HBSS) supplemented with 10 mM HEPES, sodium salt (pH 7.4) and ascorbic acid (0.1%). Excess drug was added to the buffer in a scintillation vial. After sonication for 30 min, the scintillation vials were placed on a shaker (100 rpm) in a 37°C walk-in incubator. Samples withdrawn at different time points were filtered through a 0.45 μm filter. The samples were diluted with isopropanol and stored at −20°C until further analysis. Solubility was determined from the equilibrium concentration. Fenretinide was practically insoluble in HBSS-HEPES, pH 7.4 buffer (below quantifiable limits i.e., <10 ng/mL). This necessitated the formulation of fenretinide as a solid dispersion to enhance its solubility. Fenretinide, when formulated as a Povidone K 25 solid dispersion had a solubility of more than 2.5 mg/mL.
Caco-2 cell culture
Caco-2 cells (passage: 55–78) were grown in vent capped 75 cm2 tissue culture flasks at 37°C in a humidified atmosphere of 5% CO2. DMEM supplemented with 10% FBS, 15 mM HEPES, pH 7.4, 2% L-glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin was used as the culture medium. For transport studies, cells were seeded at a density of 70,000 cells/cm2 on collagen-coated Transwell® inserts. The apical (AP) and basolateral (BL) compartments contained 1.5 and 2.5 mL of the culture medium, respectively. The cells were allowed to grow and differentiate for 18–22 days before use. The integrity of the cell monolayer was assessed before, during, and after the study by measurement of transepithelial electrical resistance (TEER) using an epithelial voltohmmeter (World Precision instruments, West Haven, CT). Monolayers with TEER >500 Ω ·cm2 were utilized for the transport studies. Antipyrine was used as a marker compound for passive transcellular transport [12].
Drug transport across Caco-2 monolayers
HBSS (pH 7.4) supplemented with 10 mM HEPES and 0.1% ascorbic acid was used as the transport medium. Transport experiments were initiated by washing the monolayers three times with HBSS followed by pre-incubation of the cell monolayers in HBSS for 30 min. Various concentrations (50–250 μg/mL or 125–640 μM) of fenretinide solutions were prepared by dissolving appropriate amounts of fenretinide solid dispersion in the transport medium. Apical to basolateral (AP-BL) and basolateral to apical (BL-AP) flux studies were performed by introducing the drug solution into the AP and BL side, respectively. Transport medium in the presence or absence of 4% BSA was added to the receiver chamber. Samples were removed from the receiver at various time points (10, 20, 40, 60, 90, 120, 150, and 180 min) over 3 h and analyzed by HPLC. The receiver was replenished with an equal volume of transport medium. Samples were also withdrawn from the donor chamber at the beginning and end of each experiment. AP-BL transport studies of fenretinide were also carried out at 0, 2, and 3% BSA or 17 μg/mL retinol binding protein (RBP) in HBSS-HEPES. For paracellular transport studies, transport medium was prepared without calcium. Permeability of fenretinide under pH gradient conditions (pH 6.0 in AP and pH 7.4 in BL) was also studied.
All experiments were conducted as triplicates and the results were expressed as mean ± SD. The statistical significance was evaluated using Student's t test.
Calculation of apparent permeability coefficient
The apparent permeability coefficient, P app (cm/sec), for all the transport studies was calculated using the following equation:
where, ΔQ/Δt is the rate of appearance of the drug in the receiver chamber (μg/min), A is the cross-sectional area (cm2) of the semi-permeable membrane of the transwell, and C 0 is the initial donor concentration (μg/mL).
The P app of fenretinide was normalized with a correction factor to account for variations from one monolayer to the other. The permeability of antipyrine, a passive diffusion marker was determined along with that of fenretinide during each transport study. The correction factor was obtained by dividing the P app of antipyrine (during a particular study) with the average P app of antipyrine obtained from control monolayers. The average P app of antipyrine was found to be 4.1 × 10−5 cm/sec by performing transport studies of antipyrine in a number of transwells (n = 8; TEER >500 Ω ·cm2) and confirmed by repeating these studies on separate days.
Extraction of fenretinide
After the transport experiments, the Caco-2 monolayers were rinsed with ice-cold PBS, scraped off, and subjected to a freeze-thaw cycle. After centrifugation, the supernatant was analyzed for the amount of drug in the cytosolic portion of the cells. The amount of drug in the lipophilic cell membrane was estimated by treating the pellet with ethanol overnight. The insert in each well was incubated with ethanol to extract the drug adsorbed to the insert.
Saturated plasma protein binding concentrations of fenretinide
The saturated protein binding concentrations of fenretinide were determined in different concentrations of BSA (0, 2, 3, and 4%), 17 μg/mL RBP (0.84 μM), and 17 μg/mL RBP + 4% BSA in HBSS-HEPES, pH 7.4. Excess amount of drug was added to the protein solutions in teflon-capped glass bottles. After sonication for 30 min, the bottles were placed on a shaker (100 rpm) at 37°C. Samples (1 mL) were withdrawn at different time points till equilibrium was achieved. The amount of fenretinide in the samples was determined after protein precipitation (with isopropanol).
HPLC analyses
Samples were analyzed by HPLC consisting of a pump (Waters 510), autosampler (Waters 717 plus), and a UV-Vis detector (Shimadzu SPD-10A). A Nova-Pak C18 3.9 × 300 mm (Waters, Milford, MA) column equipped with a C18 guard column was employed during the analysis. The samples (1.0 mL) were spiked with 50 μL of internal standard (20 μg/mL of all trans-retinol) followed by addition of isopropanol (1.0 mL) to precipitate the protein. The mixture was vortexed and centrifuged. The supernatant was then analyzed for fenretinide and antipyrine. The mobile phase used in the analysis of fenretinide comprised of acetonitrile/water/acetic acid (75:23:2) at a flow rate of 1 mL/min and wavelength of 365 nm. The calibration range for fenretinide was 0.03–100 μg/mL. The limit of quantification (LOQ) and limit of detection (LOD) were determined to be 0.03 μg/mL and 0.01 μg/mL, respectively.
Antipyrine was analyzed according to a published method at 254 nm with a mobile phase composed of acetonitrile/78 mM phosphate buffer (25:75, pH 7.2, 1 mL/min) [13].
Results and discussion
Caco-2 monolayer permeability studies
The maximum concentration of fenretinide (dissolved as solid dispersion) used in the transport studies was 250 μg/mL, which corresponds to about 0.2% w/v Povidone K 25. As the TEER across Caco-2 monolayers in the presence of up to 0.5% w/v Povidone K 25 was not significantly different than the TEER in control studies (without Povidone K 25), the cell monolayer integrity was not altered by the presence of Povidone K 25 during transport studies (Fig. 2). Also, the permeability of a passive diffusion marker, antipyrine, was not affected after treatment with 0.5% Povidone K 25 [(38.0 ± 4.0)× 10−6 cm/sec] in comparison to the control [(41.0 ± 1.8)× 10−6 cm/sec].

TEER across Caco-2 cell monolayers in the presence of 0.5% w/v Povidone K 25 in the apical (AP) compartment. The TEER values obtained at different time points are reported as percentage of TEER from control experiments (absence of Povidone K 25). Data represents mean ± SD (n = 3)
The mass balance was more than 90% in all experiments. The permeability of fenretinide in the absence of BSA in the receiver medium could not be determined due to extremely poor aqueous solubility of fenretinide. Subsequently, transport studies were performed in the presence of 4% BSA in the receiver as the albumin content in human blood is about 4 g/dL [14].
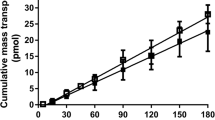
The P app of fenretinide across the insert in the absence of Caco-2 cells was several folds (>1000) higher than that in the presence of the cell monolayer. The barrier properties of the filter support membrane were therefore considered to be negligible while calculating P app. Permeability studies of different concentrations of fenretinide ranging from 50–250 μg/mL were carried out in the AP-BL direction across Caco-2 monolayers. A linear relation (Fig. 3) was observed between the flux (2.5 × 10−4 μg/min ·cm2 at 50 μg/mL to 1.3 × 10−3 μg/min·cm2 at 250 μg/mL) and the concentration of fenretinide within this concentration range, indicating a passive diffusion pathway for the transport of fenretinide across Caco-2 monolayers.

AP-BL flux of fenretinide across Caco-2 monolayers as a function of initial drug concentration. Fenretinide solution (50 to 250 μg/mL) was loaded on the AP side followed by sampling from the BL side. Data represents mean ± SD (n = 3)
The permeability of fenretinide was independent of the apical pH (6.0 or 7.4), thereby suggesting the lack of involvement of a proton-dependent transporter (Table 1). Yamashita et al. studied the effect of pH (6.0 or 7.4 in AP; 7.4 in BL) on permeability of a wide range of passively and actively transported drugs. Cephalexin and cefutibuten, known substrates of the proton-gradient dependent oligopeptide transporter (PepT1) showed significantly higher permeability under the pH-gradient condition in comparison to the permeability with a pH of 7.4 on both the AP and BL sides [15]. Also, a better prediction of the absorbed fraction in humans was reported when pH-gradient conditions were employed.
To further elucidate the type of passive diffusion pathway involved (paracellular vs. transcellular), transport studies were performed in the absence of calcium. The absence of this divalent cation tends to open up tight junctions, thereby enhancing the permeability of paracellularly transported compounds [16]. Extracellular calcium concentrations had no significant effect on the permeability of fenretinide indicating that paracellular transport is not a significant pathway for a highly lipophilic substance like fenretinide (Table 1). Also, no significant difference (p > 0.05) was observed between the apparent permeability coefficients for the AP-BL and BL-AP directions at two different concentrations (Fig. 4). Based on the lack of difference in the permeability values of fenretinide in the AP-BL and BL-AP directions, it is unlikely that an efflux transporter like P-glycoprotein (P-gp) is involved in limiting the permeability by pumping out fenretinide from the cells.

P app values for fenretinide across Caco-2 monolayers for the AP-BL and BL-AP directions. The drug solution at pH 7.4 (45 or 85 μg/mL) was loaded in the AP or BL compartment for the AP-BL and BL-AP directions, respectively. Values are represented as mean ± SD (n = 3)
Fenretinide (final concentration of 25 μg/mL) was incubated with Caco-2 cell suspension over a period of three hours (duration of the transport study) to determine its metabolism by the cells. A previous study revealed enzymatic hydrolysis of mefenamic acid-guaiacol ester by Caco-2 homogenate, which resulted in a very short half-life for the ester prodrug [17]. In our studies, the metabolism of fenretinide in Caco-2 cells was found to be negligible (<1% for 100 μg/mL after 3 h). Also, stability studies over a 24 h period in the transport medium indicated that drug degradation was minimal (<2%) during the initial three hour period. Therefore, metabolism and degradation may not limit the permeability of fenretinide across Caco-2 cells.

P app of fenretinide in the presence of 2, 3, and 4% (300–600 μM) BSA and/or 17 μg/mL (0.8 μM) RBP in the receiver (BL) compartment
Permeability of fenretinide in the presence of plasma proteins
The P app of fenretinide increased with an increase in the BSA concentration (Fig. 5). However, the permeability of antipyrine, a passive diffusion marker was similar either with 4% BSA in the receiver [(41.0 ± 1.8)×10−6 cm/sec] or without BSA [(42.0 ± 1.7)× 10−6 cm/sec]. Krishna et al. suggested that inclusion of BSA provides the necessary absorptive driving force and better mimics the in vivo sink conditions [18]. For highly lipophilic and poorly water soluble compounds like fenretinide, the presence of BSA in the receiver, apart from overcoming a compromise in sink conditions, tends to increase the permeation out of the cell membrane by drug-protein binding [15, 19]. According to Aungst et al. who carried out permeability studies of HIV protease inhibitors and other lipophilic drugs, an enhancement in permeability was observed only for the most lipophilic (log P > 3) and highly protein-bound (>95%) drugs, such as chlorpromazine, nelfinavir, and DMP 851 [20]. The data in this study is in agreement with literature as the permeability of fenretinide (log P=8.03; highly protein bound) was enhanced in the presence of BSA, whereas the permeability of antipyrine (log P=0.39; poor plasma protein binding) [21, 22] remained unchanged. However, even at a BSA concentration of 4%, fenretinide exhibited poor permeability (8.8 × 10−8 cm/sec), which might be due to poor partitioning of the highly lipophilic moiety between the cell membrane and the receiving medium. A similar observation was reported for cosalane, a highly lipophilic anti-HIV agent (log P = 6.8), which had a very low P app (4.49 × 10−8 cm/sec) in the presence of 1% BSA [23].
The maximum fenretinide concentration in different protein solutions was determined and the data is shown in Table 2. Sink conditions are maintained throughout the study as the fenretinide concentrations achieved in the receiver at any time point are well below (<15%) the maximum achievable concentrations in the corresponding protein solutions.
Although the exact mechanism of fenretinide transport in plasma is not known, it is quite likely to be bound to plasma proteins such as albumin, lipoproteins and serum retinol-binding protein (RBP). The average plasma level of fenretinide administered at a dose of 200 mg/day over a 5-year treatment period during a breast cancer prevention study was found to be about 1 μM (∼0.39 μg/mL) [24]. Owing to the extremely poor aqueous solubility of fenretinide (<10 ng/mL), the plasma concentrations achieved might be due to plasma protein binding. RBP, a 21 kDa glycoprotein forms a complex with all-trans retinol (ATRol) in the liver and is involved in the transport of ATRol in the blood [25]. Fenretinide interacts with RBP to form a tight complex, but the affinity is lower than that of retinol [26].
Transport studies of fenretinide were also carried out with RBP in the receiver due to the fact that it binds to retinoids with a greater affinity in comparison to albumin. The RBP concentration used in the receiver was 17 μg/mL (0.8 μM), similar to that in human plasma [27]. The P app of fenretinide in the presence of RBP and 4% BSA was found to be higher than that in the presence of 4% BSA alone (Fig. 5). Previously, a 14-fold increase in permeability of chlorpromazine (CPZ) was observed when 0.1% BSA was replaced with 0.1% α1-acid glycoprotein, which is known to bind to CPZ with a much greater affinity than BSA [28]. The role of an appropriate receiver solution during transport studies of molecules like CPZ, which have poor permeabilities due to accumulation in the cell membrane was emphasized. Similarly, the permeability of highly lipophilic compounds like fenretinide should depend on the receiver composition and the experimental results show this dependency (Table 2 and Fig. 5).
Accumulation of fenretinide in the cell monolayer
Lipophilic drugs are generally believed to permeate extremely well because of greater partitioning into the phospholipid bilayer of the cell membrane. However, for highly lipophilic drugs with a log P greater than 3.5, a decrease in permeability is observed probably due to a low solubility coupled with accumulation in the cell lipid bilayer [29, 30]. In this study, a large amount (∼13–15% of the initial amount) of fenretinide was found to accumulate in the cell membrane after the transport study. Lack of detectable quantities of fenretinide transport in in vitro permeation across rat everted gut sacs studies and appearance of dark yellow color (fenretinide is yellow) due to extensive accumulation of fenretinide confirmed its accumulation (data not shown). In a similar study, metabolism, paracellular transport and efflux did not contribute to the poor permeation of cosalane; instead, cosalane permeability across Caco-2 cells was limited by accumulation in the cell membrane and poor partitioning into the receiver [23]. Sawada et al. also state that poor permeability of highly lipophilic compounds is due to limited desorption from the receiver-side membrane into the buffer, which is further restricted by in vitro assays with a limited volume of receiver [31]. In accordance with lipophilic compounds like chlorpromazine [28], pyrimidine-based antioxidants [31], and cosalane [23], accumulation in the cell membrane might be playing a role in the poor permeability of fenretinide. As further proof for accumulation-limited transport of fenretinide, disappearance (from donor, AP) and appearance (in receiver, BL) studies of fenretinide across the monolayer demonstrate a distinct difference between the uptake and appearance profiles of fenretinide (Fig. 6). The amount of fenretinide in the donor decreased by 16% of the initial amount (60 μg) over a 3 h period. However, only 0.12% of the initial amount in the donor made it to the receiver. Poor partitioning between the cell membrane and the receiver compartment might also limit the permeability of fenretinide. The permeability seems to increase with an increasing amount of protein due to increased drug binding.

Uptake from the AP or donor compartment (▪) and appearance in the BL or receiver compartment (□) as a function of time are shown (A). A t is the amount of fenretinide present in the AP or BL compartment at any time t. A 0 is the initial amount in the donor (60 μg). A magnified plot of appearance in the receiver is also shown (B). A BL is the amount of fenretinide in the BL side. Receiver (BL) contained 4% BSA in HBSS, pH 7.4
Conclusions
Fenretinide permeates through the Caco-2 monolayer by transcellular passive diffusion. Based on the in vitro results, the in vivo oral absorption of fenretinide might be limited due to accumulation in the cell membrane and poor partitioning into the receiving plasma. However, solubility and dissolution of fenretinide in the gut lumen might also pose a problem as the in vivo studies employed an oil formulation and not a solid dispersion. Further absorption studies of fenretinide as solid dispersion filled in capsules in animal models may prove to be useful in elucidating the role of dissolution as a limiting factor.
References
Pollard M, Luckert PH, Sporn MB (1991) Prevention of primary prostate cancer in Lobund-Wistar rats by N-(4-hydroxyphenyl) retinamide. Cancer Res 51:3610–3611
Formelli F, Barua AB, Olson JA (1996) Bioactivities of N-(4-hydroxyphenyl) retinamide and retinoyl beta-glucuronide. Faseb J 10:1014–1024
Lieberman R, Crowell JA, Hawk ET, Boone CW, Sigman CC, Kelloff GJ (1998) Development of new cancer chemoprevention agents: role of pharmacokinetic/pharmacodynamic and intermediate endpoint biomarker monitoring. Clin Chem 44:420–427
Veronesi U, De Palo G, Costa A, Formelli F, Decensi A (1996) Chemoprevention of breast cancer with fenretinide. IARC Sci Publ 87–94
Veronesi U, De Palo G, Marubini E, Costa A, Formelli F, Mariani L, Decensi A, Camerini T, Del Turco MR, Di Mauro MG, Muraca MG, Del Vecchio M, Pinto C, D’Aiuto G, Boni C, Campa T, Magni A, Miceli R, Perloff M, Malone WF, Sporn MB (1999) Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J Natl Cancer Inst 91:1847–1856
Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, Di Mauro MG, Costa A, Marubini E, Sporn MB, De Palo G (2006) Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol 17:1065–1071
Fontana JA, Rishi AK (2002) Classical and novel retinoids: their targets in cancer therapy. Leukemia 16:463–472
Reynolds CP, Matthay KK, Villablanca JG, Maurer BJ (2003) Retinoid therapy of high-risk neuroblastoma. Cancer Lett 197:185–192
Swanson BN, Zaharevitz DW, Sporn MB (1980) Pharmacokinetics of N-(4-Hydroxyphenyl)-all-trans-retinamide in rats. Drug Metab Dispos 8:168–172
Nozawa T, Imai K, Nezu J, Tsuji A, Tamai I (2004) Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in human. J Pharmacol Exp Ther 308:438–445
Law D, Krill SL, Schmitt EA, Fort JJ, Qiu Y, Wang W, Porter WR (2001) Physicochemical considerations in the preparation of amorphous ritonavir-poly(ethylene glycol) 8000 solid dispersions. J Pharm Sci 90:1015–1025
Lennernas H (1998) Human intestinal permeability. J Pharm Sci 87:403–410
Ooie T, Terasaki T, Suzuki H, Sugiyama Y (1997) Quantitative brain microdialysis study on the mechanism of quinolones distribution in the central nervous system. Drug Metab Dispos 25:784–789
Fisher JM, Wrighton SA, Calamia JC, Shen DD, Kunze KL, Thummel KE (1999) Midazolam metabolism by modified Caco-2 monolayers: effects of extracellular protein binding. J Pharmacol Exp Ther 289:1143–1150
Yamashita S, Furubayashi T, Kataoka M, Sakane T, Sezaki H, Tokuda H (2000) Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur J Pharm Sci 10:195–204
Gan LS, Hsyu PH, Pritchard JF, Thakker D (1993) Mechanism of intestinal absorption of ranitidine and ondansetron: transport across Caco-2 cell monolayers. Pharm Res 10:1722–1725
Tantishaiyakul V, Wiwattanawongsa K, Pinsuwan S, Kasiwong S, Phadoongsombut N, Kaewnopparat S, Kaewnopparat N, Rojanasakul Y (2002) Characterization of mefenamic acid-guaiacol ester: stability and transport across Caco-2 cell monolayers. Pharm Res 19:1013–1018
Krishna G, Chen K, Lin C, Nomeir AA (2001) Permeability of lipophilic compounds in drug discovery using in-vitro human absorption model, Caco-2. Int J Pharm 222:77–89
Saha P, Kou JH (2002) Effect of bovine serum albumin on drug permeability estimation across Caco-2 monolayers. Eur J Pharm Biopharm 54:319–324
Aungst BJ, Nguyen NH, Bulgarelli JP, Oates-Lenz K (2000) The influence of donor and reservoir additives on Caco-2 permeability and secretory transport of HIV protease inhibitors and other lipophilic compounds. Pharm Res 17:1175–1180
Higaki K, Asai M, Suyama T, Nakayama K, Ogawara K, Kimura T (2002) Estimation of intradermal disposition kinetics of drugs: II. Factors determining penetration of drugs from viable skin to muscular layer. Int J Pharm 239:129–141
He YL, Tsujimoto S, Tanimoto M, Okutani R, Murakawa K, Tashiro C (2000) Effects of protein binding on the placental transfer of propofol in the human dually perfused cotyledon in-vitro. Br J Anaesth 85:281–286
Pal D, Udata C, Mitra AK (2000) Transport of cosalane-a highly lipophilic novel anti-HIV agent-across caco-2 cell monolayers. J Pharm Sci 89:826–833
Formelli F, Clerici M, Campa T, Di Mauro MG, Magni A, Mascotti G, Moglia D, De Palo G, Costa A, Veronesi U (1993) Five-year administration of fenretinide: pharmacokinetics and effects on plasma retinol concentrations. J Clin Oncol 11:2036–2042
Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ, De Luca LM (2000) Retinoids in chemoprevention and differentiation therapy. Carcinogenesis 21:1271–1279
Zanotti G, Marcello M, Malpeli G, Folli C, Sartori G, Berni R (1994) Crystallographic studies on complexes between retinoids and plasma retinol-binding protein. J Biol Chem 269:29613–29620
de Pee S, Dary O (2002) Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr 132:2895S–2901S
Sawada GA, Ho NF, Williams LR, Barsuhn CL, Raub TJ (1994) Transcellular permeability of chlorpromazine demonstrating the roles of protein binding and membrane partitioning. Pharm Res 11:665–673
Wils P, Warnery A, Phung-Ba V, Legrain S, Scherman D (1994) High lipophilicity decreases drug transport across intestinal epithelial cells. J Pharmacol Exp Ther 269:654–658
Rubas W, Cromwell M (1997) The effect of chemical modifications on octanol/water partition (log D) and permeabilities across Caco-2 monolayers. Adv Drug Deliv Rev 23:157–162
Sawada GA, Barsuhn CL, Lutzke BS, Houghton ME, Padbury GE, Ho NF, Raub TJ (1999) Increased lipophilicity and subsequent cell partitioning decrease passive transcellular diffusion of novel, highly lipophilic antioxidants. J Pharmacol Exp Ther 288:1317–1326
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kokate, A., Li, X. & Jasti, B. Transport of a novel anti-cancer agent, fenretinide across Caco-2 monolayers. Invest New Drugs 25, 197–203 (2007). https://doi.org/10.1007/s10637-006-9026-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-006-9026-3