
Abstract
Purpose
To analyse the effects of long-term memantine treatment on the retinal physiology and morphology of DBA/2J mice.
Methods
DBA/2J (D2J) mice received i.p. injections of the NMDA receptor antagonist memantine, which protects neurons from abnormally elevated glutamate levels, twice a day over a period of 7 months. At the age of 2, 6 and 10 months, the intraocular pressure (IOP) and electroretinograms (ERGs) were measured in all treated D2J mice, in untreated D2J controls and in C57Bl/6 (B6) wild-type mice. After the last measurement at the age of 10 months, the mice were killed and the retinae and the optic nerves were analysed morphologically.
Results
The IOP increased with age in both D2J and B6 mice with a larger increase in the D2J strain. IOPs were not influenced by memantine treatment. The response amplitude of the scotopic flash ERG decreased with age in the D2J strain. This amplitude decrease, particularly that of the b-wave, was smaller in treated D2J mice. The retinae of treated D2J mice exhibited less peripheral degeneration of cone photoreceptors, and optic nerve neuropathy was less frequent.
Conlcusions
Application of the NMDA receptor antagonist memantine diminished retinal neurodegeneration in the D2J mice and had a protective effect on the b-wave amplitude of the scotopic flash ERG. This protection may occur secondarily as memantine primarily acts on retinal ganglion cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The DBA/2J (D2J) mouse is a widely used mouse model for secondary angle-closure glaucoma. D2J mice carry mutations in the Gpnmb R150X and Tyrp 1b gene, coding for two melanosomal proteins [1]. As a consequence, D2J mice develop iris stroma atrophy and iris pigment dispersion with age [2, 3]. The dispersed iris pigment material blocks the outflow pathways of the aqueous humour, leading to increased intraocular pressures (IOPs) starting approximately at the age of 6 months [4–6]. The classical view is that increased IOP results in a progressive loss of retinal ganglion cells (RGCs) and optic nerve degeneration [2, 4, 7, 8]. Consistent with the structural changes, RGC function is severely compromised in older D2J mice as measured with pattern electroretinography (pERG) [9]. Moreover, the decrease of the mean pERG amplitude shows a strong correlation with the mean IOP elevation [5]. Recent full-field ERG measurements, however, show that D2J mice display additional functional deficits before the IOP increases [10]. The missing optomotor reflex in young D2J mice also indicates further functional deficits [11]. Although structural changes occur at an age at which the IOP increases, there is no strict correlation between structural [6] and functional changes [10] on the one hand and the IOP on the other hand in individual eyes. These data suggest that other neurodegenerative mechanisms are probably involved.
Glutamate excitotoxicity is considered to be a major cause of neuronal cell injury in both acute and chronic neurodegenerative diseases of the central nervous system [12–14]. The degeneration of RGCs during glaucoma is presumably also related to disturbed levels of glutamate. Already in 1957, Lucas and Newhouse [15] reported toxic effects mainly on the RGC layer of mice that received subcutaneous injections of glutamate. Olney [16] observed glutamate-induced retinal degeneration in neonatal mice and introduced the term “excitotoxicity”. In a study by Vorwerk et al. [17], chronic low-dose glutamate resulted in a significant loss of RGCs, and experimental glutamatergic excitotoxicity severely altered the spiking activity of rabbit RGCs [18].
The excitotoxicity induced by glutamate is supposed to be caused by the excessive activation of the NMDA-type glutamate receptor channel [12, 13], leading to increased intracellular Na+ and Ca2+ concentrations. Especially, disturbed intracellular Ca2+ concentrations may have serious effects, as Ca2+ is a regulator of many important cellular processes [19] and, most notably, can also trigger cell death [20, 21].
Memantine is an uncompetitive open channel blocker of NMDA receptor channels with unique kinetics and a good clinical tolerability [22–26]. It is currently used in the treatment of Parkinson’s disease [27, 28] and moderate forms of Alzheimer’s disease [29, 30]. In addition, memantine has been shown to be protective on retinal cells in several in vitro and some in vivo studies. Pellegrini and Lipton [31], for example, showed that delayed administration of memantine protected rat retinal cells in vitro after NMDA-mediated excitotoxic insult. In a rat model of pressure-induced retinal ischaemia, memantine reduced the loss of ganglion cells [32]. Hare and Wheeler [18] report that memantine blocks experimental glutamatergic excitotoxicity on rabbit RGCs.
In the D2J mouse, a structurally protective effect of memantine, that is, a significantly higher number of surviving RGCs after memantine treatment is reported by several studies [33–35], but a study of functional protective effects of memantine treatment in the D2J mouse is lacking. Moreover, it is unclear how structural and functional deficits are related to each other.
In this study, we present the results of long-term memantine treatment effects on the electroretinogram (ERG) as a physiological assay and on the structural integrity of the optic nerve and the retina.
Materials and methods
Animals
All experiments were performed in accordance with the ARVO Statements for the Use of Animals in Ophthalmic and Vision Research and were approved by the local animal welfare authorities. Six-week-old female animals were purchased from Charles River Laboratories (Sulzfeld, Germany), the European distributor of Jackson Laboratories DBA/2J mice. The mice were housed under a 12-h light and 12-h dark cycle. The mean illuminance in the housing facility was 200 lux. Food and water were provided ad libitum.
The experiments were performed on five C57Bl/6 (B6) and 20 D2J mice. All investigated B6 and five D2J individuals served as wild-type and untreated intrastrain controls, respectively. B6 mice have a different genetic background than D2J mice. Therefore, a comparison between the data from these two strains will not give information about the genotype–phenotype relationships. However, the measurements with the B6 mice can give an insight into how the structure and physiology in animals without neurodegeneration are. They further enable a comparison with literature data in which B6 mice were used. Finally, recordings with these animals may ensure that no changes occurred in the recording procedures during the extended period in which data were obtained.
The remaining 15 D2J individuals received intraperitoneal injections of memantine (5 mg/kg, memantine concentration 0.625 mg/ml, sterile and pyrogen free, dissolved in potassium sorbate and aqua ad inject) over a period of 7 months starting at the age of 3 months. The injections were given twice a day (mornings and late afternoon) on 5 days per week (weekends excluded).
At the age of 2, 6 and 10 months, we measured the IOP and recorded ERGs of all animals. After the last measurements, the animals were killed and the eyes were removed for histological preparation of the retina and the optic nerve.
IOP measurements
The IOPs of both eyes were measured between 2.00 and 3.00 p.m. under room lighting conditions (200 lux) and 1 day prior to ERG recording in all investigated animals. For non-invasive IOP measurements, we used a rebound tonometer, kindly provided by Thomas W. Mittag (Department of Ophthalmology, Mount Sinai School of Medicine, New York, NY). The calibration procedure of the tonometer is described elsewhere [37]. The impact–rebound tonometer (Tonopen; Medtronics, Jacksonville, FL) was especially developed and tested for IOP measurements in murine eyes [6, 10, 36–38]. Briefly, the animals were restrained in a custom-made device, exposing the heads of the animals for the IOP measurements without increasing the intrathoracic pressure. The measurements were performed on awake animals, thereby preventing any effects of anaesthesia on IOP. To obtain reliable data, five succeeding measurements of each eye were averaged. The left eye was measured first.
ERG measurements
A detailed description of the ERG measurements and data analysis has been given elsewhere [10]. Briefly, the animals were dark adapted over night and all further handling was performed under deep red illumination. The mice were anaesthetized by an intramuscular injection of 50 mg/kg ketamine (Ketavet®, Pfizer, Berlin, Germany) and 10 mg/kg xylazine (Rompun® 2 %, Bayer, Leverkusen, Germany). Directly after anaesthesia, the animals received a subcutaneous injection of saline solution (10 ml/kg, 0.9 %, B.Braun, Melsungen, Germany) to prevent desiccation. The pupils were dilated with a drop of tropicamide (Mydriaticum Stulln®, 5 mg/ml, Pharma Stulln GmbH, Stulln, Germany) and phenylephrin-hydrochloride (Neosynephrin POS® 5 %, Ursapharm, Saarbrücken, Germany). For the measurements, the animals were transferred onto a warming table. ERGs were measured simultaneously from the right and left eyes. The ground needle electrode was placed subcutaneously at the base of the tail, the reference needle electrodes were positioned subcutaneously on the skull medially to the ears and the active contact lens electrodes (Mayo Corporation, Japan), internally covered with Corneregel® (Dr. Mann Pharma, Berlin, Germany), were placed on the cornea of each eye. After dark adaptation for 5 min, the measurements were started. At this time, the pupils were always maximally dilated. We did not observe a difference in pupil size between animal groups nor did we observe an age-dependent change in pupil size. To deliver the stimuli, a Ganzfeld Bowl (Roland Consult Q450, Roland Consult, Brandenburg, Germany) was used. White LEDs served as light sources. The spectral output of the LEDs shows two peaks: one narrow peak with a maximum at about 445 nm and a broader second peak with a maximum at about 550 nm. Stimulation and data recording were controlled using the RetiPort system (Roland Consult, Brandenburg, Germany).
Initially, the dark-adapted flash ERG was measured. Depending on stimulus strength, between three and 12 flashes of the same strength were presented and the responses were averaged. Flash strength increased in five steps starting at 0.0003 to 0.003, 0.03, 0.3 cd.s/m2 and finally 3.0 cd.s/m2. Inter-flash intervals increased from 5 s at the lowest flash strength to 14 s at the highest flash strength. Flash duration varied between 5 μs and 5 ms depending upon the required strength.
After the measurements, the animals received an additional subcutaneous injection of saline solution and were transferred into the cage on a warming blanket where they were allowed to wake up.
ERG data analysis
For the offline analysis of the responses, we used custom-written Matlab® (©1994–2010 The Mathworks, Inc., Ismaning, Germany) routines and Excel (©1985–2003 Microsoft Corporation, Redmond, WA) spreadsheets.
The detailed procedure of the analysis of the scotopic ERG responses has been described previously [10]. Briefly, the oscillatory potentials (OPs) of the flash ERGs were separated from the other components offline by applying a fast Fourier transform to obtain the frequency spectrum of the responses. The spectra of all ERG recordings displayed two distinct amplitude regions in the frequency domain: a low-frequency region reflecting the a- and b-waves and a high-frequency region originating in the OPs (see also Zhang et al. [39]). To separate these two regions, the minimum between them was determined for each individual recording. This minimum typically was found at a frequency of about 70 Hz. Subsequently, the amplitudes above (to isolate the a- and b-wave) or below (to isolate the OPs) the minimum were set to zero. The phase plot of the Fourier spectrum was not changed. Thus, the frequency range for the ERGs without OPs was between 1 and about 70 Hz, whereas the frequency range for the OPs was between about 70 and 300 Hz. Inverse Fourier transforms were performed on the low-frequency region or high-frequency region to retrieve the ERG without the OPs or the isolated OPs, respectively.
The ERG waveforms without OPs were used to measure the amplitude and implicit time of the a- and b-wave. The a-wave amplitude was measured as the difference between the baseline level before stimulus onset and the minimum of the a-wave. The b-wave amplitude was defined as the difference between the a-wave minimum and the b-wave maximum. The amplitude of the OPs was defined as the amplitude maximum of the high-frequency part of the Fourier spectrum. To quantify age-dependent amplitude changes, the amplitudes were normalized to the values measured in the same eye at an age of 2 months. Implicit times were defined as the time between stimulus onset and the minimum of the a-wave or maximum of the b-wave, respectively. Implicit times were only included if the amplitude of the respective wave was adequately over noise level. The noise level was estimated from recordings without stimulating the retina. These recordings were treated identically as recordings with a stimulus to obtain noise estimates for the different components. Noise was about 10, 15 and 0.4 μV for the measurement of the a-waves, b-waves and OPs, respectively.
Retina whole mount preparation
The morphology and the structural integrity of the retinae and the optic nerves of B6 and D2J eyes were investigated. After the last ERG measurements at the age of 10 months, the animals were killed and the eyes were enucleated, fixed for 2 h in 4 % paraformaldehyde and rinsed overnight in PBS. Whole mount preparations were obtained by dissection of the retinae from the eyecups. Four to six small peripheral incisions were made to allow the retinae to be flat mounted on a microscope slide. The flattened retinae were treated both with Fluorescin labelled Peanut Agglutinin (PNA; fl-1071, Vector laboratories, USA) and an antibody against α-Rhodopsin (sc-20139, Santa Cruz Biotechnology, Inc., USA) according to the manufacturer’s instructions. Afterwards, the slides were washed and coverslipped (Ultra Cruz™ Mounting Medium, sc-24941, Santa Cruz Biotechnology, Inc., USA). Images were acquired using a confocal laser scanning microscope (Pascal LM5; Zeiss, Germany).
Optic nerve sections
Optic nerve neuropathy was assessed as described elsewhere [40]. In brief, optic nerves were dissected from the eye bulbs fixed in Ito’s solution and embedded in epoxy resin (Epon; Hexion Specialty Chemicals, Inc., Houston, TX). Cross sections were stained with toluidine blue. To assess the degree of optic neuropathy, areas with gliotic changes and axon degeneration in each individual eye were defined, and the percentage was calculated relative to the total cross-section area. When more than 20 % of the total cross-section area exhibited degenerative changes, the optic nerve was classified as an optic nerve with neuropathy. Photographs of optic nerve sections were taken with a light microscope (Aristoplan; Leitz, Wetzlar, Germany) equipped with a digital camera (DC500; Leica, Wetzlar, Germany).
Statistical analysis
Statistical analyses were performed with SPSS V17 (©IBM Corporation 2011, Somers, NY). Differences between two groups (untreated and memantine-treated D2J mice) were tested using the Mann–Whitney U Test, and differences between more than two groups were tested with the Kruskal–Wallis Test. Results were corrected for multiple testing. P values < 0.05 were considered statistically significant.
Results
IOP measurements
Figure 1 shows the IOPs measured in B6 wild-type controls and in untreated and memantine-treated D2J mice at the age of 2, 6 and 10 months. The IOPs of the left and right eyes are shown separately together with the mean for each group. Initially, at the age of 2 months, the B6 mice had a mean IOP of 8.4 mmHg, whereas the mean IOP of D2J mice was slightly lower (7.5 mmHg). There was no difference between the IOP of untreated and treated D2J mice at this age, indicating that there was no a priori bias in the two populations. With increasing age, the IOP increased in all mice. This increase was markedly larger in the D2J mice. At the age of 6 months, the IOPs of treated D2J mice were higher than those of untreated animals. This difference, however, was not significant and is possibly a result of the daily handling with these animals. At the age of 10 months, the mean IOP of B6 mice was 11.3 mmHg, whereas it was more than 13 mmHg in the D2J mice. This difference was significant (p < 0.05 for B6 vs. D2J untreated and p < 0.001 for B6 vs. D2J treated). The IOPs were not significantly different between untreated and treated D2J mice. The IOP data from B6 and untreated D2J mice are consistent with those from former studies [4, 5, 10, 41]. We would like to emphasize that the current study was not meant to investigate the influence of IOP on retinal degeneration. For that purpose, a comparison with mouse strains that have a similar genetic background as D2J mice without an IOP increase, such as the D2.Gpnmb+ mouse [42], would be more appropriate.

IOP measurements in B6, untreated D2J and memantine-treated D2J mice. Results of IOP measurements performed at the age of 2, 6 and 10 months in B6 (open circles), untreated (black triangles) and treated (grey squares) D2J mice. Values of the measurements in single eyes as well as the mean of each group (short dash) are shown. Initially, the IOP is slightly lower in D2J mice. With increasing age, the IOP increases in all animals. This increase is larger in D2J animals and independent of memantine treatment. At the age of 10 months, the IOP of D2J mice is significantly larger than in B6 animals (*p < 0.05 for B6 vs. D2J untreated and **p < 0.001 for B6 vs. D2J treated)
Degenerative changes in the retina and the optic nerve
The retinae of some D2J mice displayed areas with loss of cone photoreceptor cell αPNA-staining exclusively detectable in the periphery of the retina shown in Fig. 2a–c. The area with staining loss of cone photoreceptors in the periphery did in no case reach more than 10 % of the whole retinal area. In contrast, the αRhodopsin-staining pattern of rod photoreceptor cells was normal in all D2J eyes and throughout all retinal regions (central + peripheral retina) (Fig. 2d). Moreover, D2J eyes showed degenerative changes of the axons, evident by axon swelling, activated glia cells and reduced axon density within the optic nerve (Fig. 2e+f). Neither the retinae nor the optic nerves of B6 mice showed similar locally restricted areas with massive cell loss at the age of 10 months. We cannot exclude the possibility of age-related changes in overall cell density on the basis of the present study. However, it is known from the literature that no overall cell loss occurs [43].

Morphological analysis of the D2J retina and of the optic nerve. Immunofluorescence staining of cone and rod photoreceptor cells of an untreated D2J retina whole mount preparation (a–d). Fan-shaped loss of the αPNA-staining (green; cone staining) is exclusively found in peripheral regions of the retina in 50 % of the memantine-untreated and 19 % of the memantine-treated D2J eyes at the age of 10 months (red marked areas in panel A). Apparently, unstained areas next to the red markings correspond to artificial tissue folding due to the whole mount preparation. The area embraced in the white frame on the left in panel A is shown at higher magnifications in b and c. Arrows in panel B and C indicate the transition line of positive αPNA-staining to staining loss areas. d Magnification of the upper white-framed area in panel A. αRhodopsin-staining (yellow) showed a normal distribution of rod photoreceptor cells in the D2J mouse retina. The green punctate staining indicates positive αPNA-staining of cone photoreceptors in this area. Scale bars a 1 mm; b 500 μm; c, d 100 μm. e Cross section showing the morphology of an unaffected optic nerve of a 10-month-old B6 mouse. At the same age, severe optic nerve neuropathy is found in D2J mice (f). In this case, more than 80 % of the cross-section area is affected by axon loss and gliosis
A larger number of eyes of untreated D2J animals (50 %) than of memantine-treated D2J animals (19 %) displayed retinal damage as defined by αPNA-staining loss. Optic nerve neuropathy was observed in 59 % of the eyes of treated animals and in 80 % of the eyes of untreated animals. Figure 3 summarizes the results of all D2J eyes of which both the retina and the optic nerve were analysed.

Quantitative comparison of retina and optic nerve damage in treated and untreated D2J eyes. Percentage of eyes with retinal or optic nerve (ON) damage only, with both retinal and ON damage or with no damage for untreated and treated D2J mice at the age of 10 months. In memantine-treated D2J mice, fewer eyes display structural damage and the number of eyes with retinal damage is clearly reduced
Consistent with previous data, the occurrence of a structural damage was not correlated with the IOP measured in the same eye [6, 40].
Scotopic flash ERG
We previously found that the flash ERG was initially similar in B6 and D2J mice [10]. In older D2J mice (6 months and older), however, the flash ERG amplitudes decreased, possibly reflecting the onset of a damage mechanism. The major components of the flash ERG (a- and b-wave) originate in the photoreceptor cells and in second order neurons of the retina. With the flash ERG analysis, we studied whether a possible protective effect of memantine treatment on retinal ganglion cells [33–35] could also be accompanied by a functional protection revealed by a smaller age-dependent decrease of the flash ERG amplitude.
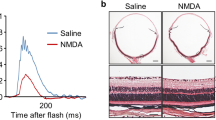
Figure 4a shows the originally recorded scotopic ERG responses measured in a B6 (left), an untreated D2J (middle) and a memantine-treated D2J (right) animal at the age of 2 months. Figure 4b shows the responses at all three investigated ages after offline filtering of the originally recorded responses as shown in Fig. 4a. In 2-month-old animals, the response amplitudes were similar in all three groups. While the responses of the B6 mice have similar amplitudes at all three ages, a reduction of the response amplitudes can be observed in the D2J animals. Particularly, a reduced a-wave is visible in the responses of 10-month-old D2J mice in comparison with the responses recorded at 2 months of age. The decrease of the b-wave amplitude is also recognizable in the responses of the untreated and treated D2J animals. This decrease is, however, slightly smaller in the treated animal. All responses display large OPs on the leading edge of the b-wave. The OPs seem to be smaller in the D2J animals but there is no obvious age-related change in any of the animals.

Scotopic flash ERG responses of B6 and D2J mice. a Original scotopic flash ERG responses measured in 2-month-old B6, untreated and treated D2J mice at a flash strength of 0.3 cd.s/m2. The vertical dashed line indicates the time of flash onset. Traces represent the mean response obtained for each group. At the age of 2 months, the responses are similar in all three groups. b Scotopic flash ERG responses at all three investigated ages after offline filtering of the originally recorded responses. The low-frequency components (a- and b-wave) were separated from the high-frequency components (OPs) using inverse fourier transforms. The obtained signals were used for the amplitude and latency measurements. In the B6 control group, the responses remain unchanged over the consecutive measurements, whereas the responses of D2J animals exhibit a more prominent amplitude reduction which is larger in the untreated D2J group
To quantify the age-dependent amplitude changes, we normalized the measured amplitudes to those measured in the same eye at the age of 2 months. The normalized amplitudes (mean + SD; error bars are shown one sided in this logarithmic plot) of B6 mice, untreated D2J controls and memantine-treated D2J mice at the age of 6 and 10 months are shown in Fig. 5 as a function of flash strength. Values smaller than one indicate an amplitude reduction relative to the first measurement, and values larger than one reflect an increase in amplitude. The amplitude reduction is similar for all flash strengths. Thus, the amplitude decrease does not depend on stimulus strength. Normalization was performed to decrease the influence of inter-individual variability. The data of the B6 mice show that in wild-type animals, the response amplitudes were not changed. In addition, the response amplitudes from individual eyes in successive measurements were correlated with the amplitudes measured at an age of 2 months (data not shown), showing that the inter-individual variability can be repeatedly measured in different sessions. This indicates that (i) the recording procedure and experimental set-up has not been changed during the extended period in which the measurements were performed, (ii) the physiology of normal wild-type animals is not altered during the first 10 months and (iii) the normalization procedure is indeed valid for reducing the influence of inter-individual variability.

Normalized scotopic ERG amplitudes of B6 and D2J mice. Normalized mean (±SD) amplitudes of the scotopic a-wave (a), b-wave (b) and the oscillatory potentials (c) of B6 mice (filled circles), D2J controls (open squares) and treated D2J mice (filled squares) at the age of 6 (left panels) and 10 months (right panels) plotted as a function of flash strength. The response amplitudes were normalized to those obtained in the first measurement at the age of 2 months. (*p < 0.01 for D2J treated vs. D2J untreated)
The relative a-wave amplitude is shown for the two highest flash strengths because at lower flash strengths no distinct a-wave was present (Fig. 5a). At the age of 6 months, the a-wave amplitude is decreased by about 40 % in both D2J groups compared to its value at 2 months of age. In contrast, the a-wave of B6 wild-type mice is not reduced. At the age of 10 months, the a-wave of B6 mice is still as large as at the age of 2 months, whereas the a-wave of D2J mice further decreased to approximately 50 % of its size at 2 months of age. Larger data sets and possibly more flash strengths would be necessary to determine whether the reduction is significant and to establish a possible cause of this reduction. There is no significant difference between the a-wave amplitude of treated and untreated D2J mice at both ages.
The relative amplitude of the b-wave measured in B6 wild-type mice did not display an age-dependent decrease (Fig. 5B). Similarly, the b-wave amplitude of 6-month-old memantine-treated D2J mice is as large as at the age of 2 months, whereas the b-wave of the untreated D2J cohort is reduced by 20 % on average at this age. At the age of 10 months, the b-wave amplitude of treated D2J animals is reduced by approximately 20 % and the b-wave amplitude of untreated D2J mice decreased by approximately 40 % of its original value. Statistical analysis revealed a significant difference between the b-wave of untreated and memantine-treated D2J mice (p < 0.001), for both age groups. The mean b-wave amplitude is 20 % larger in treated than in untreated D2J mice at the age of 10 months.
The normalized OP amplitudes are shown in Fig. 5c. With increasing age, B6 animals display OPs of equal amplitude as those measured at the age of 2 months. In contrast, the OP amplitude of D2J mice was reduced by about 30 % at the age of 6 months and by about 20 % at the age of 10 months. At an age of 6 months, the OPs in the treated animals were significantly smaller than those of untreated animals. At the age of 10 months, the OP amplitudes did not differ significantly between the two groups.
In addition to the amplitude, we analysed the implicit times of the scotopic a- and b-wave (data not shown). We found that the implicit times were similar for all groups and at all ages.
Correlation between morphological and physiological data
Furthermore, we studied whether the morphological changes in the retina (αPNA-staining loss) and the optic nerve were correlated with the changes of the flash ERG amplitude. We compared the normalized b-wave amplitudes (average of the normalized amplitudes measured at the two highest flash strengths) at the age of 10 months in eyes that displayed optic nerve degeneration with the amplitudes measured in eyes without optic nerve degeneration. Similarly, the normalized b-waves of eyes with and without cone photoreceptor cell degenerations were compared (Fig. 6). This comparison was performed separately for treated and untreated animals.

Normalized b-wave amplitude in relation to retinal and optic nerve damage in D2J mice. Normalized b-wave amplitudes (±SD) of untreated (gre y bars) and treated (black bars) D2J mice at the age of 10 months. The normalized response amplitudes at the two highest flash strengths (0.3 and 3.0 cd.s/m2) were averaged for each eye and then grouped depending on the results of the morphological examination of the retinae and the optic nerves, respectively. The b-wave amplitude is always less reduced in treated D2J mice, but there is no difference between eyes with or without retinal or optic nerve (ON) damage, respectively
The relative b-wave amplitudes were consistently larger in memantine-treated than in untreated mice, even though this difference never reached significance (Fig. 6). The b-wave amplitude reduction measured in eyes with a retinal damage compared to eyes without a retinal damage showed also no significant difference, neither for the treated nor for the untreated group. Similarly, there was no significant difference between eyes with and without an optic nerve damage.
Discussion
Flash ERG
The dark-adapted flash ERG is normal in D2J mice at an age of 2 months. However, the amplitude of the flash ERG decreases with increasing age. The recent data of Harazny et al. [10] show that the age of decrease onset coincides with IOP increase. However, we confirm previous data that the functional deficits are not correlated with IOP elevation in the same eye [6, 8, 10]. In addition, we previously found that the photopic flicker ERG displays deficits that precede the IOP changes and the changes in the scotopic flash ERG, suggesting a second (early) type of functional deficit. This is possibly in agreement with behavioural data [11].
We found that memantine treatment had a small but detectable protective effect against the functional degenerations. The scotopic b-wave amplitude was approximately 20 % larger in memantine-treated D2J mice in comparison with the untreated group (Fig. 5b). This difference was significant. The b-wave is thought to originate in bipolar cell activity [44]. This indicates that these degenerations apparently also affect outer retinal structures. Hare et al. [45] found that memantine has functional protective effects in monkeys with experimental glaucoma as measured with the multifocal ERG. In contrast, in the same study, all components of the conventional flash ERG were unaffected in experimental glaucoma irrespective of whether the animals were treated by memantine or by the vehicle. This indicates that the retinal degeneration in the D2J mouse and in experimental glaucoma in monkeys probably do not involve completely identical pathological processes.
Memantine is a blocker of open NMDA channels and therefore protective against glutamate excitotoxicity that is believed to play a role in glaucomatous retinal degeneration. Experimentally, memantine-reversed short-term excitotoxic effects induced via the NMDA receptor channel on RGC spiking activity in the rabbit retina [18]. The flash ERG was not altered. In the present experiment, we found that long-term treatment with memantine can slow down a retinal degeneration. Even though the glaucomatous damage in the D2J mouse may primarily affect the retinal ganglion cells, it cannot be excluded that secondarily cells in the outer retina are involved. This may explain the protective effect of memantine treatment on the b-wave and on the photoreceptor cells (see below). Based on the present study, it is not possible to establish unequivocally whether the effects are caused by memantine or by the handling procedure, for which a comparison with vehicle-treated animals would be more appropriate. We have not injected the untreated animals with the vehicle in the present study, because we initially wanted to establish that the D2J data were comparable with previous data.
Here, we used the scotopic flash ERG to compare the physiology of the different animal groups. The flash ERG, however, mainly originates in the outer retina. It has been shown that the pattern ERG (pERG) has more inner retinal origins and may even to a large extent reflect ganglion cell activity [46, 47]. Indeed, it has been shown that the pERG is substantially altered in the D2J mouse [5, 9, 48]. The pERG might provide a better physiological assay to establish the physiological effect of memantine treatment.
Morphological changes
Structurally, D2J eyes exhibited degeneration of the optic nerve and loss of cone photoreceptor cells (revealed by a loss of αPNA-staining). The changes of the optic nerve have been described before [2, 8, 40]. The cone-specific αPNA-staining loss was found to be present in patches located in the retinal periphery comparable to previous findings on age-dependent retinal ganglion cell loss in retinae of D2J and DBA/2NNia mice [8, 49]. This is consistent with the above-mentioned idea that the photoreceptoral degenerations possibly occur secondarily to the changes in the inner retina. Of 22 D2J eyes, both the retina and the optic nerve were morphologically investigated and in only one of these eyes the retinal damage was not accompanied by degeneration of the optic nerve. In contrast, in ten D2J eyes, the optic nerve was damaged without a retinal degeneration, suggesting that the damage of the optic nerve occurs prior to the retinal damage. Of the 22 eyes that were examined morphologically, 14 came from seven animals and therefore left and right eyes were investigated. The remaining eight eyes came from animals of which only one eye was included in the study. Of the seven animals of which both eyes were studied morphologically, only three eyes showed retinal damage. All three came from different animals. Optic nerve damage was observed unilaterally in six animals and only one animal displayed a damage in both optic nerves. This indicates that there was no obvious correlation between damage in left and right eyes of the same animal, and therefore, we treated them as independent samples.
If it is true that photoreceptor damage occurs after damage of the inner retina, it can be expected that the patches with loss of αPNA-staining might underlie larger areas with inner retinal damage. It is known that RGCs are lost regionally in the D2J retina [8, 49, 50] and that the degeneration of the axons within the optic nerve is similarly regionalized [51]. Interestingly, these regions were also described as being fan shaped like our observed peripheral cone loss. This further supports the statement that, at a given point in time, photoreceptors suffer secondarily, probably in those areas where RGC axons and RGCs themselves were affected earlier. The protective effect of memantine on photoreceptors may therefore also be secondarily through the protective effects on the RGCs.
A structural protection of memantine treatment in the D2J retina has been shown by several studies [33–35], which all report an increased number of surviving ganglion cells. Consistent with these results, we found that retinal damage was more abundant (50 %) in untreated animals than in treated animals (19 %). The fraction of eyes that displayed optic nerve damage differed by approximately 20 % between memantine-treated (59 %) and untreated (80 %) animals. This suggests that memantine treatment might possibly slow down a retinal damage by partially protecting the optic nerve and/or slowing its degeneration.
In contrast to Schuettauf et al. [52], who showed that photoreceptor cells are ultrastructurally unaffected in the D2J retina, we found that at least cone photoreceptor cells thoroughly might degenerate with age. This is consistent with latest results of our group concerning ultrastructural examinations of rod and cone photoreceptor ribbon synapses which show that the typical synaptic structure of the photoreceptor ribbon synapse is altered in the ageing D2J mouse (Scholz, et al. IOVS 2011;52: ARVO E-Abstract 2452).
The data presented in this study strongly suggest that morphological damage and functional deficits are probably related to each other. Thus, it is tempting to speculate that any treatment protecting a tissue from neurodegenerative processes may also have positive physiological and thus functional protective effects.
References
Anderson MG, Smith RS, Hawes NL, Zabaleta A, Chang B, Wiggs JL, John SWM (2002) Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat Genet 30(1):81–85
John SWM, Smith RS, Savinova OV, Hawes NL, Chang B, Turnbull M, Roderick TH, Heckenlively JR (1998) Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci 39(6):951–962
Chang B, Smith RS, Hawes NL, Anderson MG, Zabaleta A, Savinova O, Roderick TH, Heckenlively JR, Davisson MT, John SW (1999) Interacting loci cause severe iris atrophy and glaucoma in DBA/2J mice. Nat Genet 21(4):405–409. doi:10.1038/7741
Libby RT, Anderson MG, Pang IH, Robinson ZH, Savinova OV, Cosma IM, Snow A, Wilson LA, Smith RS, Clark AF, John SWM (2005) Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci 22(5):637–648
Saleh M, Nagaraju M, Porciatti V (2007) Longitudinal evaluation of retinal ganglion cell function and IOP in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 48(10):4564–4572
Scholz M, Buder T, Seeber S, Adamek E, Becker CM, Lütjen-Drecoll E (2008) Dependency of intraocular pressure elevation and glaucomatous changes in DBA/2J and DBA/2J-Rj mice. Invest Ophthalmol Vis Sci 49(2):613–621
Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH (2005) Changes in retinal populations in the DBA/2J mouse. Cell Tissue Res 320(1):51–59
Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW (2006) Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci 7:66
Porciatti V, Saleh M, Nagaraju M (2007) The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci 48(2):745–751
Harazny J, Scholz M, Buder T, Lausen B, Kremers J (2009) Electrophysiological deficits in the retina of the DBA/2J mouse. Doc Ophthalmol 119(3):181–197
Barabas P, Huang W, Chen H, Koehler CL, Howell G, John SW, Tian N, Renteria RC, Krizaj D (2011) Missing optomotor head-turning reflex in the DBA/2J mouse. Invest Ophthalmol Vis Sci 52(9):6766–6773. doi:10.1167/iovs.10-7147
Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1(8):623–634
Meldrum B, Garthwaite J (1990) Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 11(9):379–387
Lipton SA, Rosenberg RA (1994) Mechanisms of disease: excitatory amino acids as a final common pathway in neurologic disorders. N Engl J Med 330(9):613–622
Lucas DR, Newhouse JP (1957) The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 58(2):193–201
Olney JW (1969) Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol 28(3):455–474
Vorwerk CK, Lipton SA, Zurakowski D, Hyman BT, Sabel BA, Dreyer EB (1996) Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest Ophthalmol Vis Sci 37(8):1618–1624
Hare WA, Wheeler L (2009) Experimental glutamatergic excitotoxicity in rabbit retinal ganglion cells: block by memantine. Invest Ophthalmol Vis Sci 50(6):2940–2948
Clapham DE (1995) Calcium signaling. Cell 80(2):259–268
Schanne FA, Kane AB, Young EE, Farber JL (1979) Calcium dependence of toxic cell death: a final common pathway. Science 206(4419):700–702
Trump BF, Berezesky IK (1995) Calcium-mediated cell injury and cell death. FASEB J 9(2):219–228
Bormann J (1989) Memantine is a potent blocker of N-methyl-D-aspartate (NMDA) receptor channels. Eur J Pharmacol 166(3):591–592
Kornhuber J, Bormann J, Retz W, Hubers M, Riederer P (1989) Memantine displaces [3H]MK-801 at therapeutic concentrations in postmortem human frontal cortex. Eur J Pharmacol 166(3):589–590
Seif el Nasr M, Peruche B, Rossberg C, Mennel HD, Krieglstein J (1990) Neuroprotective effect of memantine demonstrated in vivo and in vitro. Eur J Pharmacol 185(1):19–24
Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA (1992) Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci 12(11):4427–4436
Lipton SA (1993) Prospects for clinically tolerated NMDA antagonists: open-channel blockers and alternative redox states of nitric oxide. Trends Neurosci 16(12):527–532
Schneider E, Fischer PA, Clemens R, Balzereit F, Fünfgeld EW, Haase HJ (1984) Effects of oral memantine administration on Parkinson symptoms. Results of a placebo-controlled multicenter study. Dtsch Med Wochenschr 109(25):987–990. doi:10.1055/s-2008-1069311
Greenamyre JT, O’Brien CF (1991) N-methyl-D-aspartate antagonists in the treatment of Parkinson’s disease. Arch Neurol 48(9):977–981
Orgogozo JM, Rigaud AS, Stoffler A, Mobius HJ, Forette F (2002) Efficacy and safety of memantine in patients with mild to moderate vascular dementia: a randomized, placebo-controlled trial (MMM 300). Stroke 33(7):1834–1839
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348(14):1333–1341. doi:10.1056/NEJMoa013128
Pellegrini JW, Lipton SA (1993) Delayed administration of memantine prevents N-methyl-D-aspartate receptor-mediated neurotoxicity. Ann Neurol 33(4):403–407. doi:10.1002/ana.410330414
Lagreze WA, Knorle R, Bach M, Feuerstein TJ (1998) Memantine is neuroprotective in a rat model of pressure-induced retinal ischemia. Invest Ophthalmol Vis Sci 39(6):1063–1066
Schuettauf F, Quinto K, Naskar R, Zurakowski D (2002) Effects of anti-glaucoma medications on ganglion cell survival: the DBA/2J mouse model. Vision Res 42(20):2333–2337
Zhong L, Bradley J, Schubert W, Ahmed E, Adamis AP, Shima DT, Robinson GS, Ng YS (2007) Erythropoietin promotes survival of retinal ganglion cells in DBA/2J glaucoma mice. Invest Ophthalmol Vis Sci 48(3):1212–1218. doi:10.1167/iovs.06-0757
Ju WK, Kim KY, Angert M, Duong-Polk KX, Lindsey JD, Ellisman MH, Weinreb RN (2009) Memantine blocks mitochondrial OPA1 and cytochrome c release and subsequent apoptotic cell death in glaucomatous retina. Invest Ophthalmol Vis Sci 50(2):707–716. doi:10.1167/iovs.08-2499
Filippopoulos T, Matsubara A, Danias J, Huang W, Dobberfuhl A, Ren L, Mittag T, Miller JW, Grosskreutz CL (2006) Predictability and limitations of non-invasive murine tonometry: comparison of two devices. Exp Eye Res 83(1):194–201
Goldblum D, Kontiola AI, Mittag T, Chen B, Danias J (2002) Non-invasive determination of intraocular pressure in the rat eye. Comparison of an electronic tonometer (TonoPen), and a rebound (impact probe) tonometer. Graefes Arch Clin Exp Ophthalmol 240:942–946
Danias J, Kontiola AI, Filippopoulos T, Mittag T (2003) Method for the noninvasive measurement of intraocular pressure in mice. Invest Ophthalmol Vis Sci 44(3):1138–1141
Zhang K, Yao G, Gao Y, Hofeldt KJ, Lei B (2007) Frequency spectrum and amplitude analysis of dark- and light-adapted oscillatory potentials in albino mouse, rat and rabbit. Doc Ophthalmol 115(2):85–93
Birke MT, Neumann C, Birke K, Kremers J, Scholz M (2010) Changes of osteopontin in the aqueous humor of the DBA/2J glaucoma model correlated with optic nerve and RGC degenerations. Invest Ophthalmol Vis Sci 51(11):5759–5767. doi:10.1167/iovs.10-5558
Inman DM, Sappington RM, Horner PJ, Calkins DJ (2006) Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest Ophthalmol Vis Sci 47(3):986–996
Howell GR, Libby RT, Marchant JK, Wilson LA, Cosma IM, Smith RS, Anderson MG, John SW (2007) Absence of glaucoma in DBA/2J mice homozygous for wild-type versions of Gpnmb and Tyrp1. BMC Genet 8:45
Samuel MA, Zhang Y, Meister M, Sanes JR (2011) Age-related alterations in neurons of the mouse retina. J Neurosci 31(44):16033–16044. doi:10.1523/jneurosci.3580-11.2011
Stockton RA, Slaughter MM (1989) B-wave of the electroretinogram: a reflection of ON bipolar activity. J Gen Physiol 93(1):101–122
Hare WA, WoldeMussie E, Lai RK, Ton H, Ruiz G, Chun T, Wheeler L (2004) Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey I: functional measures. Invest Ophthalmol Vis Sci 45(8):2625–2639
Porciatti V (2007) The mouse pattern electroretinogram. Doc Ophthalmol 115(3):145–153. doi:10.1007/s10633-007-9059-8
Miura G, Wang MH, Ivers KM, Frishman LJ (2009) Retinal pathway origins of the pattern ERG of the mouse. Exp Eye Res 89(1):49–62. doi:10.1016/j.exer.2009.02.009
Nagaraju M, Saleh M, Porciatti V (2007) IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Invest Ophthalmol Vis Sci 48(10):4573–4579
Danias J, Lee KC, Zamora MF, Chen B, Shen F, Filippopoulos T, Su Y, Goldblum D, Podos SM, Mittag T (2003) Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57BL/6 mice. Invest Ophthalmol Vis Sci 44:5151–5162
Jakobs TC, Libby RT, Ben Y, John SWM, Masland RH (2005) Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J Cell Biol 171(2):313
Howell GR, Libby RT, Jakobs TC, Smith RS, Phalam FC, Barter JW, Barbay JM, Marchant JK, Nagaraju M, Porciatti V, Whitmore AV, Masland RH, John SWM (2007) Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol 179(7):1523–1537
Schuettauf F, Rejdak R, Walski M, Frontczak-Baniewicz M, Voelker M, Blatsios G, Shinoda K, Zagorski Z, Zrenner E, Grieb P (2004) Retinal neurodegeneration in the DBA/2J mouse-a model for ocular hypertension. Acta Neuropathol 107(4):352–358. doi:10.1007/s00401-003-0816-9
Acknowledgments
The work was supported by Novartis Institutes for BioMedical Research. JK is supported by the Excellence Program of the Hertie Foundation. MS is supported by the Johannes und Frieda Marohn Foundation, Erlangen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jenny Atorf, Michael Scholz contributed equally to this work.
Rights and permissions
About this article
Cite this article
Atorf, J., Scholz, M., Garreis, F. et al. Functional protective effects of long-term memantine treatment in the DBA/2J mouse. Doc Ophthalmol 126, 221–232 (2013). https://doi.org/10.1007/s10633-013-9380-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10633-013-9380-3