
Abstract
Background
Pancreatic ductal adenocarcinoma (PDA) has a poor prognosis due to its therapeutic resistance. Inactivation of vitamin D/vitamin D receptor (VDR) signaling may contribute to the malignant phenotype of PDA and altered expression of oncoprotein mucin 1 (MUC1) may be involved in drug resistance of cancer cells.
Aim
To determine whether vitamin D/VDR signaling regulates the expression and function of MUC1 and its effect on acquired gemcitabine resistance of pancreatic cancer cells.
Methods
Molecular analyses and animal models were used to determine the impact of vitamin D/VDR signaling on MUC1 expression and response to gemcitabine treatment.
Results
RPPA analysis indicated that MUC1 protein expression was significantly reduced in human PDA cells after treatment with vitamin D3 or its analog calcipotriol. VDR regulated MUC1 expression in both gain- and loss-of-function assays. Vitamin D3 or calcipotriol significantly induced VDR and inhibited MUC1 expression in acquired gemcitabine-resistant PDA cells and sensitized the resistant cells to gemcitabine treatment, while siRNA inhibition of MUC1 was associated with paricalcitol-associated sensitization of PDA cells to gemcitabine treatment in vitro. Administration of paricalcitol significantly enhanced the therapeutic efficacy of gemcitabine in xenograft and orthotopic mouse models and increased the intratumoral concentration of dFdCTP, the active metabolite of gemcitabine.
Conclusion
These findings demonstrate a previously unidentified vitamin D/VDR-MUC1 signaling axis involved in the regulation of gemcitabine resistance in PDA and suggests that combinational therapies that include targeted activation of vitamin D/VDR signaling may improve the outcomes of patients with PDA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer, primarily pancreatic ductal adenocarcinoma (PDA), is currently the third leading cause of cancer-related death in the USA and is projected to become the second leading cause of cancer-related death by 2030. The incidence of PDA is increasing [1]. PDA has a dismal prognosis, with a median survival duration of 8 months from diagnosis and a 5-year survival rate of less than 10% for all stages [1, 2], which is largely due to its late diagnosis and therapeutic resistance. The most effective treatment for PDA is curative resection followed by chemotherapy, but most PDA patients are unable to undergo surgical treatment because the PDA cells have disseminated to nearby tissues or distant organs by the time of diagnosis. Therefore, cytotoxic chemotherapy is the primary treatment modality for most PDA patients; however, the development of chemoresistance is extremely common during the course of treatment [3]. New treatments that are effective in other malignancies, such as checkpoint immunotherapy, have not shown efficacy in PDA. Thus, new intervention strategies are urgently needed to improve the outcomes of patients with PDA.
Gemcitabine (dFdC or 2′,2′-difluoro-deoxycytidine) is a deoxycytidine analog that is active through incorporation of its triphosphate (dFdCTP) into DNA, leading to inhibition of DNA replication and arrest of tumor growth [4]. Gemcitabine has been widely used as an anti-cancer chemotherapeutic agent for various solid tumors, including PDA. Gemcitabine plus nab-paclitaxel is the standard first-line treatment option for locally advanced and metastatic pancreatic cancer [5, 6], and gemcitabine continues to be a common component of PDA chemotherapy. Nevertheless, gemcitabine does not provide a significant clinical patient survival benefit because tumors frequently develop resistance within weeks of onset of treatment. A large number of studies have attributed this lack of response to poor penetration of gemcitabine into the hypo-vascularized and extensively desmoplastic PDA tumors [7]; however, it could not fully explain why tumors initially respond to gemcitabine treatment. Little is known, however, about the mechanisms by which tumor cells acquire gemcitabine resistance.
Vitamin D is a fat-soluble vitamin that is naturally present in foods, available as a dietary supplement, and produced endogenously in the skin after exposure to sunlight. Through binding to the vitamin D receptor (VDR) transcription factor, the biologically active form of vitamin D, 1,25-dihydroxyvitamin D3 (vitamin D3), has pleiotropic functions beyond its activity as a hormone regulating calcium phosphate homeostasis, including suppression of cell proliferation, induction of cell differentiation and apoptosis, and regulation of the microbiome and immune response [8]. The effects of vitamin D may differ during early and late stages of tumorigenesis (affecting efficacy as a preventive agent) and its effects on later stage and metastatic disease alone or in combination with other drugs remains to be clinically determined. Studies suggest that vitamin D may have survival benefits in patients with PDA [9,10,11]. Additionally, in a genome-wide screen of genes associated with overall survival, VDR was identified as a novel determinant of survival in patients with advanced pancreatic cancer. Patients who carried the rs2853564 VDR variant in G/G allele survived longer; and the interaction between this specific VDR variant with high pre-treatment levels of 25-dihydroxyvitamin D or with gemcitabine treatment was associated with longer overall survival [10]. VDR has been reported to be a master transcriptional regulator of pancreatic stellate cells, blocking their activation, reducing fibrosis, and increasing intratumoral gemcitabine and its activation resulted in a reduction in tumor volume in a mouse model of pancreatic cancer [12]. VDR activation and photodynamic priming enabled durable low-dose FDA-approved nanoliposomal irinotecan chemotherapy without compromising tumor control in mouse model of pancreatic cancer [13]. We previously found that activation of VDR signaling downregulates the expression of nuclear FOXM1 protein and suppresses pancreatic cancer cell stemness and tumorigenesis [14]. These findings suggest the potential application of activation of vitamin D/VDR signaling as a component of PDA therapy. It remains unknown whether vitamin D/VDR signaling plays a role in gemcitabine resistance particularly in acquired gemcitabine resistance in PDA cells and whether activation of vitamin D/VDR signaling can sensitize pancreatic cancer cells to gemcitabine treatment or reverse gemcitabine resistance.
Mucin 1 (MUC1), also known as epithelial membrane antigen (EMA), encodes a transmembrane glycosylated protein. The alpha and beta subunits of this protein form a heterodimeric complex: the N-terminal alpha subunit functions in cell adhesion and the C-terminal beta subunit is involved in cell signaling. In normal epithelial cells, MUC1 is highly glycosylated and tightly aligned on the apical surface, forming a mucous barrier to protect cells and maintain homeostasis. Cellular transformation is associated with loss of polarity, and underglycosylated MUC1 is overexpressed on the cell surface, often associated with growth factor receptors, which facilitates growth signal transduction and the aggressiveness of cancer cells [15, 16]. In the pancreas, MUC1 is expressed in the apices of centroacinar cells, intercalated and intralobular ducts, and focally in the interlobular ducts in normal pancreatic tissue [17]. Overexpression of MUC1 has been observed in intraepithelial neoplasia lesions, with a subsequent increase in expression associated with disease progression in human and in mouse models of pancreatic cancer [18,19,20]. Muc1 knock-out mice demonstrate slower tumor progression and fewer metastases compared with muc1 wild-type KrasG12D; P48-Cre mice [21], whereas transgenic expression of human MUC1 enhances tumor progression and immune suppression [20], defining MUC1 as an oncoprotein. MUC1 was also reported to be involved in pancreatic cancer drug resistance through regulation of multidrug resistance gene expression, affecting glucose metabolism and dCTP biosynthesis [22,23,24]. It remains unclear how MUC1 is dysregulated in acquired gemcitabine resistance and whether targeting altered MUC1 expression can improve the sensitivity of gemcitabine-resistant pancreatic cancer cells to gemcitabine treatment.
In the present study, we sought to determine whether vitamin D/VDR signaling regulates the expression and function of MUC1 in gemcitabine-resistant pancreatic cancer cells. We demonstrate that gemcitabine-resistant pancreatic cancer cells often exhibit reduced VDR but increased MUC1 expression and that activation of vitamin D/VDR signaling negatively regulates MUC1 expression and sensitizes pancreatic cancer cells to gemcitabine treatment. The results of this study provide new insights into the mechanisms of gemcitabine resistance, support a beneficial effect of vitamin D as an adjunct to treatment of patients with pancreatic cancer and suggest that vitamin D or its analogs could play a role in PDA therapy.
Materials and Methods
Cell Lines and Cell Culture
Human pancreatic cancer cell lines AsPC-1 (RRID:CVCL_0152), Capan-2 (RRID:CVCL_0026), CFPAC-1 (RRID:CVCL_1119), PANC-1 (RRID:CVCL_0480), HPAC (RRID:CVCL_3517), Hs 766 T (RRID:CVCL_0334), Mia PaCa-2 (RRID:CVCL_0428), and mouse 266-6 (RRID:CVCL_3481) acinar cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). PaTu 8902 (RRID:CVCL_1845) cell line was purchased from DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). COLO 357 (RRID:CVCL_0221) and MDA-Panc-28 (RRID:CVCL_3917) cell lines were described previously [25]. Mouse PDA cell lines PKC-mL3 and PKC-mL1 were obtained from David Tavion’s laboratory at Cold Spring Harbor Laboratory. PKC-118 is a cell line derived from the PKC mouse model. Human pancreatic stellate cell (HPSC-TERT) (RRID:CVCL_SA58) line was a gift from Rosa F. Hwang at The University of Texas MD Anderson Cancer Center. All of these cell lines were maintained in 5% CO2 at 37 °C as an adherent monolayer in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. Additionally, the following components were added to DMEM as supplements: nonessential amino acids, a multi-vitamin solution (Flow Laboratories), sodium pyruvate, and l-glutamine. The immortalized normal human pancreatic ductal epithelial HPDE-1/E6E7 (RRID:CVCL_S973) cell line (obtained from Dr. Tsao, Ontario Cancer Institute, Toronto, ON, Canada) was maintained in keratinocyte serum-free medium supplemented with epidermal growth factor and bovine pituitary extract (Invitrogen). All human cell lines were authenticated using STR profiling within the last 6 months (available on request) and all experiments were performed with mycoplasma-free cells.
Transient Transfection of Plasmids and siRNAs
JetPRIME reagent (Polyplus, Life Technologies) was used to transfect plasmid DNA or siRNA oligos according to the manufacturer’s instructions. Briefly, plasmid DNA or siRNA was diluted and mixed well in jetPRIME buffer; JetPRIME reagent was then added to the DNA or siRNA solution and mixed by vortexing and briefly spinning down. The transfection mixture was added to single-cell suspensions of pancreatic cancer cells and then plated into wells of 96- or 6-well plates for continuing growth. Cell samples were processed for analysis according to different purposes as described.
Antibodies, Plasmids, siRNAs, and Compounds
Anti-VDR (#12,550), Anti-FoxM1 (#5436), and anti-GLUT1 (#12,939) antibodies were purchased from Cell Signaling Technology. Anti-MUC1 monoclonal antibody (clone E29, #M0613) was purchased from Dako, while a second anti-MUC1 antibody (clone CT2, #MA511202) and Ki67 Ab (#14-5698-82) were purchased from Invitrogen. Anti-CD44 (SC-9960) antibody was purchased from Santa Cruz Biotechnology. VDR expression plasmid was purchased from Origene. siRNAs targeting VDR and MUC1 were purchased from Dharmacon. Gemcitabine was purchased from Eli Lilly. Standard gemcitabine triphosphate (dFdCTP) was purchased from Toronto Research Chemicals. 1,25-dihydroxyvitamin D3 and calcipotriol were purchased from Sigma. Paricalcitol was manufactured by AbbVie, Inc.
Western Blot Analysis
Standard Western blotting was done using whole-cell lysates, primary antibodies listed in 2.3, and peroxidase-linked, species-specific, anti-rabbit, anti-mouse, anti-goat, or anti-hamsterIgG F(ab’)2 fragments. For the detection of MUC1 protein, two monoclonal antibodies that recognize epitopes of MUC1 (clones E29 and CT2) with advanced verification were used to facilitate the detection of multiple markers while using the same blot membrane for internal control proteins (GAPDH, α-tubulin, or β-actin). Protein bands were detected using the Pierce™ enhanced chemiluminescence system. Protein bands were quantitated using ImageJ software; protein expression was normalized to GAPDH, α-tubulin, or β-actin, and the ratio to control was presented under individual blots as fold changes.
Quantitative Real-Time PCR (qRT-PCR) Assay
Total RNA was extracted from cultured or treated cells using the RNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions. SuperScript IV First-Strand Synthesis System (Invitrogen) was used to reversely transcribed messenger RNAs into cDNAs. Real-time PCR was performed on a StepOnePlus Real-Time PCR System (ABI) in triplicate using the Taqman method. The PCR primers and probe sets purchased from Life Technologies were as follows: VDR (Hs01045843_m1), MUC1 (Hs00904316_g1), HPRT1 (Hs99999909_m1), and β-actin (4352341E). The relative quantitation of gene expression was determined using the comparative threshold cycle (ΔΔCt) method and normalized to HPRT1 or ACTB (β-actin gene) and a calibrator sample that was run on the same plate.
In Vitro Cell Viability Assay
Human or mouse PDA cells were seeded into 96-well plates with 3000 cells per well. The drugs (gemcitabine, calcipotriol, or paricalcitol) or vehicle control were diluted in DMEM medium to indicated concentrations supplemented with 5% FBS, and 200-μL DMEM with drug or vehicle control was added to each well 12 h after cell plating. Cells were cultured at 37 °C for indicated times as described in individual experiments. After incubation, Cell Counting Kit-8 (Dojindo Molecular Technologies) was used to determine the cell viability, and the IC50 of each drug was calculated using Prism 8 software (GraphPad Software, Inc.). The viability of the vehicle control treated cells was set as 100%. Cell viability treated at each drug concentration was assayed in triplicate. In some experiments, cells in suspension were transfected with MUC1 siRNA or control siRNA mediated by JetPRIME reagent and seeded into 96-well plates. DEME Medium combined with gemcitabine or vehicle control was added to each well at 12 h after cell plating and incubated for the indicated times in related experiments. Cell viability and IC50 were determined as described above.
Reverse-Phase Protein Array
AsPC-1 cells treated with vitamin D3 (0, 0.1 µM/L) or calcipotriol (0, 0.1, 1.0 µM/L) for 48 h were harvested, and cell lysates were subjected to reverse-phase protein array (RPPA) analysis according to the standard protocol at the Functional Proteomics RPPA Core Facility at MD Anderson Cancer Center, as described previously [26]. Heatmaps of the differentially expressed proteins were generated by ‘pheatmap’ package in R4.0.3. Information about the RPPA antibodies is available at https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/antibody-information-and-protocols.html. The RPPA data were deposited into TCPA database with accession number #TCPA00000010.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed using EZ-ChIP kit (Cat#17-371, Millipore) according to the manufacturer’s instructions and as described previously [27]. Control IgG or anti-VDR antibody was used for immunoprecipitation. The resulting immunocomplexes were purified and used as templates for qPCR amplification of three fragments in the MUC1 promoter region with PowerUp SYBR Green Master 34 Mix (Applied Biosystems) using the corresponding primers as described in Supplementary Table S1.
Immunofluorescence Assay
Cells grown in Falco four-well chambered cell culture slides were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min. Fixed cells were then blocked by incubation with 5% bovine serum albumin (BSA) solution containing 0.2% Triton X-100 for 30 min. After briefly washing with PBST once, diluted VDR antibody (1:500) and MUC1 antibody (1:1000) were mixed in 5% BSA solution and incubated with cells overnight at 4 °C. After three washes with PBST, cells were incubated with Alexa Fluor 594-labeled or Alexa Fluor 488-labeled secondary antibodies (Thermo Fisher Scientific) for 2 h at room temperature. Cells were washed, mounted with prolong gold antifade reagent with DAPI (Thermo Fisher Scientific), viewed, and photographed under a fluorescence microscope (Leica).
Immunohistochemical Analysis
VDR, MUC1, and Ki67 expression in pancreatic tumor tissues were analyzed. The pancreatic tissue samples were purchased from US Biomax, Inc. (Derwood, MD) or obtained from xenograft and orthotopic mouse models of pancreatic cancer and their use was approved by the Institutional Review Board of MD Anderson Cancer Center. Tissue sections (4 µm thick) of formalin-fixed, paraffin-embedded tumor specimens were deparaffinized in xylene and rehydrated in graded alcohol. Standard immunohistochemical procedures were performed on the tissue sections using anti-VDR, anti-MUC1, or anti-Ki67 antibody. A positive reaction was indicated by brown membrane, cytoplasmic, and/or nuclear staining, depending on the antibody used. The staining result was determined by the percentage of positive cells and staining intensity and scored as described previously [28].
Xenograft Mouse Model of Human Pancreatic Cancer
Pathogen-free athymic BALB/c nude mice were purchased from the National Cancer Institute. The animals were maintained in MD Anderson animal facilities, which are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International in accordance with the current regulations and standards of the U.S. Department of Agriculture and Department of Health and Human Services. The mouse experiments were approved by the Institutional Animal Care and Use Committee. Established gemcitabine-resistant AsPC-1 cells (AsPC-1-GemR) in exponential growth phase were prepared for inoculation, and 100-μL single-cell suspensions (1 × 106 cells) in Hank’s balanced salt solution were injected subcutaneously into the abdominal flanks of the mice.
Orthotopic Mouse Pancreatic Tumor Model
C57BL/6 J mice were anesthetized with isoflurane, and a small incision (< 5 mm) was made in the abdomen to allow access to the spleen and pancreas. The pancreas was retracted through the incision, and gemcitabine-resistant mouse PKC-118 cells (100 μL, 4 × 105 cells) were injected into the pancreas within a period of 10 s using a 28-gauge needle. The pancreas was then reinserted into the abdominal cavity, the muscle layer sutured, and the skin stapled.
Drug Treatment of Tumor-Bearing Mice
Tumor-bearing xenograft and orthotopic mice were randomly divided into four groups: PBS control, gemcitabine alone (20 mg/kg in PBS), paricalcitol alone (2.5 μg/kg in PBS), and combined treatment (gemcitabine plus paricalcitol in PBS). Beginning on the third day after tumor inoculation, 100-μL PBS or drug solutions was injected intraperitoneally once every 3 days for 29 days. At the end of the experiment, the tumor-bearing mice were euthanized, and the tumors were removed and weighed. Tumor tissues were further processed for related analyses.
Analysis of dFdCTP by Ion Chromatography Mass Spectrometry
To determine the relative abundance of intracellular dFdCTP in tumor tissue samples, extracts were prepared and analyzed by ultra-high-resolution mass spectrometry at the Metabolomics Core Facility using previously published methods [29]. Briefly, tumor tissue samples collected from the mice within 2 h after the last gemcitabine and/or paricalcitol injection were flash frozen in liquid nitrogen. Approximately 30 to 100 mg of tissue was then homogenized using the Precellys tissue homogenizer (Bertin Instruments). Metabolites were extracted using 1-mL ice-cold 0.1% ammonium hydroxide in 80/20 (v/v) methanol/water. Extracts were centrifuged at 17,000×g for 5 min at 4 °C, and supernatants were transferred to clean tubes and evaporated to dryness under nitrogen.
The dried extracts were reconstituted in deionized water, and 5 μL was injected into the spectrometer for analysis. Ion chromatography (IC) mobile phase A (MPA; weak) was water, and mobile phase B (MPB; strong) was water containing 100-mM KOH. A Thermo Scientific Dionex ICS-5000 + IC system that included a Thermo IonPac AS11 column (4 µm particle size, 250 × 2 mm) kept at 30 °C was used. The autosampler tray was chilled to 4 °C. The mobile phase flow rate was 360 µL/min, and the gradient elution program was 0–5 min, 1% MPB; 5–25 min, 1–35% MPB; 25–39 min, 35–99% MPB; 39–49 min, 99% MPB; and 49–50, 99–1% MPB. To improve sensitivity, methanol was delivered by an external pump and combined with the eluent via a low dead volume mixing tee. A Thermo Orbitrap Fusion Tribrid Mass Spectrometer was used to acquire data in the ESI-negative ionization mode.
Raw data files were imported into Thermo Trace Finder software for final analysis. The relative abundance of metabolite was normalized by tissue weight.
Statistical Analysis
All in vitro and in vivo experiments were performed in triplicate or as indicated. The data from independent experiments were presented as mean ± SD, mean ± SEM, or as indicated otherwise. Statistical significance was determined by the Student t test or one-way ANOVA depending on the experiment. Data were analyzed using SPSS Statistics 24 (IBM, Armonk, New York) or GraphPad Prism 9.0 (GraphPad Software, La Jolla, CA). All significance was defined at the P < 0.05.
Results
Identification of MUC1 as a Vitamin D3/VDR Downstream Protein in Pancreatic Cancer Cells by RPPA Analysis
To elucidate the molecular mechanisms of vitamin D’s anti-tumor effects in pancreatic cancer, we performed an unbiased RPPA analysis using protein samples extracted from human pancreatic cancer AsPC-1 cells treated with vitamin D3 or different doses of calcipotriol, a potent and nonhypercalcemic vitamin D analog [30], to activate VDR signaling for 48 h. RPPA analysis identified 112 and 91 differentially expressed proteins (P < 0.05) affected by vitamin D3 and calcipotriol treatment, respectively (Fig. 1A, B), and among them 66 proteins overlapped between the two treatments (Fig. 1C), indicating that vitamin D3 and calcipotriol treatment affected similar signaling pathways. MUC1 was identified as one of the proteins significantly downregulated by vitamin D3 or calcipotriol treatment (Fig. 1D), with quantitative results presented in Fig. 1E and F.

Identification of MUC1 as a vitamin D3/VDR downstream protein in pancreatic cancer cells by RPPA analysis. Protein samples extracted from AsPC-1 cells after vitamin D3 (VD3) or calcipotriol (Cal) [0.1 μM/L (Cal-L) or 1.0 μM/L (Cal-H)] treatment for 48 h were used for RPPA analysis. Heatmaps (A, B) and Venn diagram (C) of the differentially expressed proteins (P < 0.05 by t test) for Ctr vs VD3, Ctr vs Cal-L, and Cal-H samples are also shown in (B). P values distribution of the proteins in Ctr vs VD3 and Ctr vs Cal-L comparison, thresholds of P = 0.05 are marked and MUC1 protein is highlighted in (D). MUC1 protein was significantly downregulated by the treatments, with quantitative data (Normalized log2 values) presented in (E and F). **P < 0.01, and ****P < 0.0001 vs vehicle Ctr
Validation of RPPA Findings and Demonstration of a Vitamin D-VDR Positive Signaling Loop
To further validate the RPPA findings, we examined the effects of vitamin D3 and calcipotriol treatment on MUC1 expression by Western blot analysis using the same antibody (E29) as in the RPPA assay. Significant inhibition of MUC1 protein expression by vitamin D3 or calcipotriol was observed not only in AsPC-1 cells but also in MIA PaCa-2, PaTu 8902, and HPAC pancreatic cancer cells. MUC1 is highly polymorphic and several alternatively spliced variants exist, accounting for multiple bands on Western blotting [16] (Fig. 2A–C). Paricalcitol, another vitamin D analog, is currently used to prevent or treat hyperparathyroidism in patients with chronic kidney diseases [31]. We found that paricalcitol treatment dose dependently inhibited MUC1 protein expression in AsPC-1 and COLO 357 cells (Fig. 2D). Similarly, vitamin D3, calcipotriol, and paricalcitol treatment also significantly inhibited MUC1 expression in PKC-118 mouse PDA cells (Fig. 2E). Interestingly, we found that treatment with vitamin D3 or its analogs significantly induced both endogenous and exogenous VDR protein expression in several pancreatic cells tested (Fig. 2C–G). These results suggest that vitamin D-VDR signaling activation may also increase VDR protein stability and a positive feedback loop between vitamin D and VDR signaling exists in pancreatic epithelial cells. We also found that treatment with vitamin D3 or its analogs significantly inhibited the expressions of CD44, FoxM1, and GLUT1 (Fig. 2H). CD44 and FoxM1 molecules play critical roles in the regulation of pancreatic cancer stemness and aggressiveness [25, 28, 32], while elevated GLUT1 expression is essential for oncogenic KRAS-mediated anabolic glucose metabolism, a hallmark of PDA and required for biosynthesis of nucleotide to support pancreatic tumor [33, 34]. In addition, vitamin D3 or calcipotriol treatment or VDR gene transduction significantly inhibited the growth of pancreatic cancer cells in vitro (Fig. S1A-F). Together, these results provide a molecular basis for using vitamin D3 or its analog(s) for PDA therapy.

Validation of the RPPA finding and demonstration of vitamin D3-VDR positive signaling loop in pancreatic cancer cells. Western blot analysis of MUC1 expression in AsPC-1 cells treated with vehicle, VD3 (0.1 µM), or Cal at the indicated concentrations (µM) for 48 h (A). The effects of VD3 or Cal treatment on MUC1 and/or VDR protein expression in MIA PaCa-2, PaTu 8902, and HPAC cells were also examined by Western blot assay (B, C) (note: MUC1 ab:E29 in A–C). Effect of vitamin D3 analog paricalcitol (PA) treatment (0, 75, 150 nM/L, for 48 h) on MUC1 and VDR protein expression in AsPC-1 and COLO 357 cells by Western blot assay (D). Western blot analysis of MUC1 and VDR expression in PKC-118 mouse PDA cells at 48 h after vehicle control, VD3 (0.1 μM/L), Cal (0.1 μM/L), or PA (0.1 μM/L) treatment (E). Cells were transfected with control pcDNA3.1 or Flag-tagged VDR expression vector (1.5 µg/well, 6-well plate), followed by Cal (0, 0.1 μM/L) or PA (0, 0.1 μM/L) treatment at 12 h after transfection. Protein samples were extracted at 48 h after treatment and used for Western blot analysis of exogenous Flag-VDR and/or endogenous VDR expression in mouse 266–6 pancreatic acinar cells and PKC-118 PDA cells (F) or in human pancreatic cancer AsPC-1 cells (G). Western blot analysis of VDR, CD44, FoxM1, MUC1, and GLUT1 protein expressions in AsPC-1 cells at 48 h after the indicated treatments (H) (note: MUC1 ab:CT2 in D-E, H; also refer to Figs. 3A and 5G). *P < 0.05 and ****P < 0.0001 vs vehicle Ctr
Correlation Between VDR and MUC1 Expression in Human Pancreatic Tissue Samples
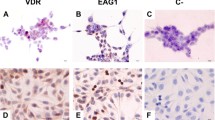
Previous studies have shown that vitamin D-VDR signaling is inactive in pancreatic cancer cells [11, 14, 35]. Altered and elevated MUC1 expression has been found in several types of cancer including pancreatic cancer, but the clinical significance and underlying molecular mechanisms between VDR and MUC1 dysregulation in PDA remain unclear. We therefore performed IHC analyses of VDR and MUC1 expression in consecutive tissue microarray (TMA) sections containing human pancreatic cancer tissue samples (Fig. S2A). Analysis of the quantitative staining results showed a negative correlation between VDR and MUC1 expression (Fig. S2B). We also found that VDR expression tended to decrease or be lost with increasing histological PDA grade, whereas MUC1 expression was in the opposite direction (Fig. S2C, D) (representative images are shown in Fig. S2E). These findings are in keeping with the function of VDR and MUC1 in regulating differentiation of cancer cells [36, 37]. We did not observe significant association of VDR/MUC1 expression with tumor stage (Tables S2-S3), but the sample size was small and the majority of the cases were early stage.
Regulatory Effects of Vitamin D/VDR Signaling on MUC1 Expression in Pancreatic Cancer Cells
To causally define the regulatory role of VDR in MUC1 expression, we first examined the expression of MUC1 and VDR protein in a panel of pancreatic cancer cell lines and found that pancreatic cancer cells differed in their expression of VDR, but that reduced or lost VDR expression tended to be associated with higher levels of MUC1, while normal pancreatic epithelial cells had no or very low levels of MUC1 expression (Fig. 3A, B). This was in line with TMA analysis (Fig. S2) and further confirmed by additional IHC analyses using both human and mouse pancreatic tissues (Fig. S3).

Regulatory effects of vitamin D3/VDR signaling on MUC1 expression in pancreatic cells. Differential expression of MUC1 and VDR protein in a panel of human pancreatic cancer cell lines (A Note: two different MUC1 antibodies recognized different epitopes and showed similar MUC1 expression patterns) and in mouse pancreatic cancer cells and human normal pancreatic cells (B). The effects of VDR gene transduction (C, D) or VDR siRNA (0, 25, 100 pM/well, 6-well plate) knockdown (E, F) on MUC1 expression in pancreatic cancer cells were determined by Western blot assay. Rescue experiment was performed in MDA-Panc-28 cells by transfection of control or VDR siRNA and followed by VD3 treatment (0, 0.1 μM/L) at 12 h after transfection. Protein samples were extracted at 48 h after transfection for Western blot analysis of VDR and MUC1 expression (G). qRT-PCR analysis of MUC1 mRNA expression in MDA-Panc-28 cells at 48 h after VDR gene transduction (H) or receiving VD3 (0.1 μM/L) or Cal (0.1 μM/L) treatment (I). Bioinformatic analysis predicted 3 potential VDR binding sites in the MUC1 promoter region (J) and measured by chromatin immunoprecipitation-quantitative PCR in AsPC-1 cells at 48 h after transfection of VDR gene expressing vector or VDR siRNA (K, L). Data presented as Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001
Next, we performed gain- and loss-of-function assays. We found that overexpression of VDR dose dependently inhibited MUC1 expression in both human and mouse pancreatic cancer cells (Fig. 3C, D), whereas knockdown of VDR expression by siRNA resulted in increased MUC1 expression (Fig. 3E, F), which could be reversed by vitamin D3 treatment (Fig. 3G). Similarly, transduction of the VDR gene or treatment of cells with vitamin D3 or calcipotriol significantly reduced MUC1 expression at the mRNA level (Fig. 3H–I). Given that VDR is a nuclear transcription factor, we performed bioinformatic analysis and identified three potential VDR binding sites in the MUC1 promoter region (Fig. 3J). The results of ChIP analysis using the cellular samples transfected with VDR-expressing vector and siRNA, respectively, indicated that VDR overexpression or knockdown differentially affected the binding of VDR protein to the VDR response elements in the MUC1 promoter region and that binding site number 2 may play a more important role in the regulation of MUC1 expression (Fig. 3K, L). Together, these results suggest that VDR plays an important role in the regulation of MUC1 expression and that activation of vitamin D-VDR signaling significantly inhibits MUC1 expression through a transcriptional mechanism.
Acquired Gemcitabine-Resistant Pancreatic Cancer Cells Exhibit Reduced VDR and Increased MUC1 Expression
Acquired gemcitabine resistance is one of the main reasons for the lack of chemotherapeutic response and poor prognosis of patients with pancreatic cancer. To explore the relationship between acquired gemcitabine resistance and altered VDR and MUC1 expression in pancreatic cancer cells, we first established two acquired gemcitabine-resistant pancreatic cancer cell lines, AsPC-1-GemR and COLO 357-GemR, by culturing the relatively sensitive AsPC-1 and COLO 357 cells in cell culture medium containing gemcitabine and gradually increasing the gemcitabine concentration for a period of greater than 4 months (Fig. 4A). Compared with their parental cells (AsPC-1-Ctr), the AsPC-1-GemR cells (Fig. 4B) displayed altered morphology, with pile-up growth in two-dimensional culture (Fig. 4C, right panel) and exhibited significantly reduced VDR but increased MUC1 protein expression, as determined by Western blot analysis (Fig. 4D). Immunofluorescent staining revealed that relatively high levels of VDR protein expression, predominantly located in the nuclear compartment, in the parental cells, which exhibited relatively low MUC1 expression. In sharp contrast, decreased expression of VDR protein distributed in both the cytoplasm and nucleus characterized AsPC-1-GemR cells, along with significantly increased MUC1 staining predominantly distributed in the cytoplasm (Fig. 4E). We also found that VDR knockdown by siRNA transfection reduced the sensitivity of AsPC-1 cells to Gem treatment (Fig. S4), again suggesting that inactivation of VDR signaling contributes to Gem resistance. Both vitamin D3 and calcipotriol treatments significantly induced VDR and suppressed MUC1 expression in AsPC-1-GemR cells (Fig. 4F), suggesting a molecular basis for using vitamin D3 or its analogs to treat gemcitabine-resistant pancreatic cancer cells. Similar results were observed in COLO 357-Ctr and -GemR cells, with the differences in VDR and MUC1 protein expressions more striking between the control and Gem-resistant cells (Fig. 4G–K).

Acquired gemcitabine-resistant pancreatic cancer cells display altered MUC1 and VDR protein expression. Diagram illustrating the approach to establish acquired gemcitabine-resistant pancreatic cancer cells by exposing the originally sensitive AsPC-1 and COLO 357 cells to an increasing concentration of gemcitabine for a period of over 4 months (A). IC50 assay of the sensitivities of parental (AsPC-1-Ctr) and acquired gemcitabine-resistant (AsPC-1-GemR) AsPC-1 cells exposed to different doses of gemcitabine for 72 h (B); representative images of AsPC-1-Ctr and AsPC-1-GemR cells (C). Western blot analysis of VDR and MUC1 protein expression in AsPC-1-Ctr and AsPC-1-GemR cells (D). Immunofluorescent staining of VDR and MUC1 protein expression and subcellular distribution in AsPC-1-Ctr and AsPC-1-GemR cells (E). Western blot analysis of VDR and MUC1 protein expression in AsPC-1-GemR cells treated with different doses of Cal and VD3 for 48 h (F). Similarly, COLO 357 cells acquired gemcitabine resistance after long-time exposure to increasing concentrations of gemcitabine in culture medium (G); representative images of COLO 357-Ctr and COLO 357-GemR cells (H). Western blot analysis of VDR and MUC1 protein expression in COLO 357-Ctr and COLO 357-GemR cells (I) and in COLO 357-GemR cells treated with VD3 (0.01 µM/L) or Cal (0.1 µM/L) (J) or with indicated doses of PA (K) for 48 h
Sensitization of Pancreatic Cancer Cells to Gemcitabine by Vitamin D Analog Treatment
Next, we determined whether activation of VDR signaling can improve the sensitivity of pancreatic cancer cells to gemcitabine treatment and performed IC50 analysis of gemcitabine with or without vitamin D analog treatment in several pancreatic cancer cell lines. We found that sensitivity to gemcitabine increased almost tenfold in MIA PaCa-2, MDA-Panc-28, PaTu 8902, and PKC-118 cells when it was combined with calcipotriol (Fig. 5A–D) and by more than twofold in established COLO 357-GemR and AsPC-1-GemR cells treated with calcipotriol or paricalcitol, respectively (Fig. 5E, F). To further understand the mechanism by which VDR signaling sensitizes pancreatic cancer cells to gemcitabine treatment, we used MUC1 siRNA to knock down MUC1 expression (Fig. 5G) and found that MUC1 siRNA transduction significantly increased sensitivity to gemcitabine treatment in COLO 357-GemR and PaTu 8902 cells (Fig. 5H, I). When the cells were treated with both MUC1 siRNA and paricalcitol, the sensitivity to gemcitabine treatment was not substantially improved compared with paricalcitol or MUC1 siRNA alone in AsPC-1-GemR cells (Fig. 5J). These results suggest that while the action of vitamin D and its analog may be exerted on MUC1, the hormone may also act by additional mechanisms in Gem-resistant PDA cells.

Activation of VDR signaling sensitizes the response of pancreatic cancer cells to gemcitabine. IC50 analysis of cell sensitivities to gemcitabine without or with Cal (0.1 µM/L) or PA (0.1 µM/L) treatment for 72 h in MIA PaCa-2 (A), MDA-Panc-28 (B), PaTu 8902 (C), PKC-118 (D), AsPC-1-GemR (E), and COLO 357-GemR (F) cells. Western blot analysis of the effect of MUC1 siRNA transfection on MUC1 and VDR protein expression in COLO 357-GemR cells (G). IC50 analysis of the sensitivities to gemcitabine treatment in cells transfected with control siRNA or MUC1 siRNA followed by gemcitabine treatment for 48 h (H, I). IC50 analysis of the sensitivities to gemcitabine in AsPC-1-GemR cells transfected with control siRNA or MUC1 siRNA followed with vehicle control or PA (0.1 µM/L) treatment for 72 h (J)
Enhanced Suppression of Tumorigenicity by Gemcitabine When Combined with a VDR Agonist
Next, we established a xenograft mouse model of human pancreatic cancer by subcutaneous injection of acquired gemcitabine-resistant AsPC-1-GemR cells into nude mice. Tumor-bearing mice were received four different treatments: vehicle control, gemcitabine or paricalcitol alone, or gemcitabine and paricalcitol combination, as depicted in Fig. 6A. Compared with the control group, the combination treatment significantly inhibited tumor growth (P = 0.016), while treatment with gemcitabine or paricalcitol alone was associated with only modest tumor growth inhibition that did not reach statistical significance (Fig. 6B, C). Single or combined treatments were not associated with significant changes in body weight or systemic toxicities (Fig. S5). Consistently, the expression of VDR was significantly induced, while expression of MUC1 and Ki67 was significantly reduced, as determined by immunohistochemical staining, in tumor tissues treated with gemcitabine and paricalcitol combination treatment compared with single-agent therapy (Fig. 6D, E). To mimic human disease, gemcitabine-resistant PKC-118 cells were used to establish an orthotopic model of pancreatic cancer in syngeneic C56BL/6 J mice, and the tumor-bearing mice were treated as described above (see Fig. 6A). In keeping with the previous experiment, the combination of gemcitabine and paricalcitol had better therapeutic efficacy than paricalcitol or gemcitabine alone (Fig. 6F, G). Immunohistochemical analysis confirmed that in the tumor tissues from mice treated with combination therapy, Ki67 staining was significantly reduced compared with single-agent treatment (Fig. 6H, I). LC-Mass Spectrometry analysis of PKC-118 tumor tissues demonstrated that the intratumoral concentration of dFdCTP was increased by more than twofold in the gemcitabine and paricalcitol combination group compared with gemcitabine treatment alone (Fig. 6J), suggesting that paricalcitol improves gemcitabine metabolism in tumor tissues.

Activation of VDR signaling enhances the therapeutic efficacy of gemcitabine in animal model of pancreatic cancer. Diagram of establishing pancreatic tumor model and treatment strategies (A). AsPC-1-GemR cells were used to establish a xenograft mouse model of human pancreatic cancer, and tumor-bearing mice received various treatments. Representative tumor images upon necropsy (B) and average tumor weights (C). Representative images of IHC staining for VDR, MUC1, and Ki67 protein expression in tumor tissue sections derived from AsPC-1-GemR xenograft tumors (D) and quantitative staining scores (E). Relatively gemcitabine-resistant mouse PKC-118 cells were used to establish an orthotopic model of pancreatic cancer in syngeneic mice, followed by different treatments, with representative pancreatic tumor images upon necropsy (F) and average tumor weights (G). Representative images of IHC staining for Ki67 in PKC-118 tumor tissue sections (H) and the quantitative staining scores (I). Measurement of intratumoral concentrations of gemcitabine triphosphate (dFdCTP) by LC–MS/MS from Gem- and Gem + PA-treated mice within 2 h after the final dose of gemcitabine; tumors from vehicle control mice served as negative controls in the assay (J). Quantitative data presented as Mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and NS no statistical difference vs Ctr
Discussion
Despite tremendous efforts, the 5-year survival rate for pancreatic cancer has not changed substantially for decades, in part because of resistance to therapy of this cancer. Understanding the molecular mechanisms underlying resistance is necessary for the development of novel and effective therapeutic modalities to improve the outcomes of patients with PDA. In the present study, we made the following findings. First in an unbiased RPPA analysis, MUC1, which was previously reported to be involved in gemcitabine resistance in pancreatic cancer, was identified as a downstream target gene of vitamin D3 and calcipotriol. Second we demonstrated that VDR negatively correlates with MUC1 expression in human pancreatic cancer tissues and regulates MUC1 expression in PDA cells through a transcriptional mechanism. Third acquired gemcitabine-resistant pancreatic cancer cells exhibit increased MUC1 and decreased VDR protein expression, and VDR ligand/agonist treatment leads to significant upregulation of VDR and downregulation of MUC1 protein expression. Fourth VDR signaling activation with a known agonist significantly enhances the sensitivity of pancreatic cancer cells to gemcitabine treatment, and suppression of MUC1 expression plays an important role in the potentiation of gemcitabine sensitivity by the VDR agonist. Fifth the combination of gemcitabine with a VDR agonist significantly inhibits tumor growth in vivo and is associated with an increased intratumoral concentration of gemcitabine bioactive metabolite. Taken together, our in vitro and in vivo data suggest the following model of vitamin D3/VDR-MUC1 signaling in gemcitabine resistance in pancreatic cancer (Fig.S6), activation of vitamin D3/VDR signaling may be a promising approach to enhance therapeutic efficacy or reverse gemcitabine resistance in patients with pancreatic cancer.
Gemcitabine-based chemotherapy continues to be a standard therapy for patients with unresectable pancreatic tumors, but most of these tumors eventually develop resistance to gemcitabine [38, 39]. The molecular mechanisms underlying acquired gemcitabine resistance remain to be determined, but several lines of evidence have linked poor therapeutic response and poor prognosis of pancreatic cancer with dysregulated vitamin D-VDR signaling [9,10,11]. Pancreatic cancer is characterized by severe desmoplasia, which is believed to be responsible for its resistance to gemcitabine therapy. A recent study suggested that VDR-mediated signaling can negatively regulate inflammation and fibrosis in pancreatitis; it can also alter tumor stroma, which resulted in increased intratumoral gemcitabine and decreased tumor volume in a mouse model of pancreatic cancer [12]. The presence of desmoplastic stroma cannot, however, fully explain why initially sensitive pancreatic cancer cells acquire gemcitabine resistance within weeks of onset of gemcitabine treatment [39]. Our finding that gemcitabine-resistant pancreatic cancer cells exhibit significantly reduced VDR and increased MUC1 protein expression compared to parental cells prompted us to speculate that tumor-cell-intrinsic mechanisms likely play a dominant role in acquired gemcitabine resistance. This hypothesis is supported by the failure of clinical trials aiming at stromal depletion despite initially promising experimental studies [40,41,42,43]. Our results also provide direct evidence that altered VDR and MUC1 expression is associated with pancreatic cancer gemcitabine resistance and agree with previous findings that VDR expression is reduced or lost in pancreatic cancer cells and that insufficient serum levels of 25-hydroxyvitamin D in patients with pancreatic cancer is associated with poor prognosis [9, 11, 14]. Interestingly, our results showed that gemcitabine-resistant pancreatic cancer cells with reduced VDR expression may still be responsive to VDR ligand treatment, resulting in upregulation of VDR and downregulation of MUC1 protein expression, indicating intact positive regulation of VDR by its ligands in pancreatic cancer cells [44], suggesting that vitamin D supplementation or VDR agonist treatment may still be relevant to pancreatic cancer patients whose tumors have reduced VDR expression. Other factors that affect vitamin D/VDR signaling and tumor features should also be considered, including the status of tumor differentiation, severity of desmoplastic reaction, level of CYP24A1 expression, and the status of p53 mutation and VDR SNPs [8]. Several clinical trials that include high-dose vitamin D3 or a VDR agonist in combination with gemcitabine-based chemotherapy for pancreatic cancer treatment are ongoing [8]. It is anticipated that vitamin D, a proven safe dietary supplement, may be a cost-effective adjunct to current cancer chemotherapy.
Currently, the molecular mechanisms leading to the downregulation of VDR in pancreatic cancer cells with acquired gemcitabine resistance remain unknown. Since mutation and rearrangement of the VDR gene are rare during carcinogenesis [45] and gemcitabine-resistant pancreatic cancer cells are still responsive to vitamin D3 and VDR agonist treatment, epigenetic mechanisms such as promoter methylation may be responsible for reduced or lost VDR expression in acquired gemcitabine resistance in pancreatic cancer cells, further elucidating the underlying mechanisms that may inform the development of strategies utilizing targeted and effective activation of VDR signaling for pancreatic cancer intervention.
Accumulating evidence suggests that altered expression of the MUC1 oncoprotein may affect many aspects of tumor biology, which has significant implications for the treatment of pancreatic cancer. MUC1 plays an important role in pancreatic carcinogenesis and progression through promoting cell proliferation, epidermal to mesenchymal transition, invasion, metastasis, and immune suppression [16, 46, 47], and siRNA-mediated suppression of MUC1 resulted in the suppression of the growth and metastasis of PDA cells [48]. MUC1 was found to be involved in the regulation of multiple metabolic pathways, particularly glycolytic metabolism, and to confer resistance of pancreatic cancer cells to gemcitabine and radiation treatment [24, 49, 50]. Clinical and experimental studies indicate that MUC1 is a promising diagnostic marker and therapeutic target for pancreatic cancer [51,52,53]. Despite the initiation of numerous clinical trials, however, the clinical benefit of targeting altered MUC1 is unproven and limited by a lack of knowledge about the mechanisms underlying dysregulated MUC1 expression. In the present study, we found, for the first time, that vitamin D3 and VDR agonist treatment negatively regulates MUC1 expression in pancreatic cancer cells and, consequently, can sensitize these cells to gemcitabine treatment, leading to enhanced suppression of tumorigenesis in mouse models when combined with gemcitabine treatment. The results from this study provide a rationale for the ongoing clinical trials using VDR agonists in combination with gemcitabine for pancreatic cancer treatment [8].
In summary, our findings indicate that dysregulated vitamin D/VDR-MUC1 signaling plays an important role in the gemcitabine resistance of pancreatic cancer and that vitamin D or VDR agonist treatment significantly downregulates MUC1 expression, enhancing the therapeutic efficacy of gemcitabine. This provides new insight into vitamin D/VDR’s anti-tumor actions and a rationale for the development of vitamin D/VDR-based combinatorial strategies for pancreatic cancer intervention.
Data Availability
The raw protein RPPA data generated in this study are available in TCPA database under accession number #TCPA00000010 (with link: http://tcpaportal.org/tcpa/download/TCPA00000010.zip). Other data that support the findings of this study are available from the corresponding author upon request.
Abbreviations
- MUC1:
-
Mucin 1
- PDA:
-
Pancreatic ductal adenocarcinoma
- PanIN:
-
Pancreatic intraepithelial neoplasia
- VDR:
-
Vitamin D receptor
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33.
Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet (London, England) 2020;395:2008–2020.
Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in pancreatic cancer. Int J Mol Sci 2019;20:4504.
Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017;9:157.
Burris HA 3rd, Moore MJ, Andersen J et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol Off J Am Soc Clin Oncol 1997;15:2403–2413.
Tsujimoto A, Sudo K, Nakamura K et al. Gemcitabine plus nab-paclitaxel for locally advanced or borderline resectable pancreatic cancer. Sci Rep 2019;9:16187.
Neesse A, Michl P, Frese KK et al. Stromal biology and therapy in pancreatic cancer. Gut 2011;60:861–868.
Wei D, Wang L, Zuo X, Bresalier RS. Vitamin D: promises on the horizon and challenges ahead for fighting pancreatic cancer. Cancers 2021;13:2716.
Cho M, Peddi PF, Ding K et al. Vitamin D deficiency and prognostics among patients with pancreatic adenocarcinoma. J Transl Med 2013;11:206.
Innocenti F, Owzar K, Jiang C et al. The vitamin D receptor gene as a determinant of survival in pancreatic cancer patients: genomic analysis and experimental validation. PLoS ONE 2018;13:e0202272.
Yuan C, Qian ZR, Babic A et al. Prediagnostic Plasma 25-hydroxyvitamin D and pancreatic cancer survival. J Clin Oncol Off J Am Soc Clin Oncol 2016;34:2899–2905.
Sherman MH, Yu RT, Engle DD et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014;159:80–93.
Anbil S, Pigula M, Huang HC et al. Vitamin D receptor activation and photodynamic priming enables durable low-dose chemotherapy. Mol Cancer Ther 2020;19:1308–1319.
Li Z, Jia Z, Gao Y et al. Activation of vitamin D receptor signaling downregulates the expression of nuclear FOXM1 protein and suppresses pancreatic cancer cell stemness. Clin Cancer Res Off J Am Assoc Cancer Res 2015;21:844–853.
Kufe DW. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis 2020;41:1173–1183.
Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med 2014;20:332–342.
Nagata K, Horinouchi M, Saitou M et al. Mucin expression profile in pancreatic cancer and the precursor lesions. J Hepato-Biliary-Pancreat Surg 2007;14:243–254.
Hinoda Y, Ikematsu Y, Horinochi M et al. Increased expression of MUC1 in advanced pancreatic cancer. J Gastroenterol 2003;38:1162–1166.
Rachagani S, Torres MP, Kumar S et al. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J Hematol Oncol 2012;5:68.
Tinder TL, Subramani DB, Basu GD et al. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J Immunol (Baltimore, Md: 1950) 2008;181:3116–3125.
Besmer DM, Curry JM, Roy LD et al. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res 2011;71:4432–4442.
Nath S, Daneshvar K, Roy LD et al. MUC1 induces drug resistance in pancreatic cancer cells via upregulation of multidrug resistance genes. Oncogenesis 2013;2:e51.
Ren J, Agata N, Chen D et al. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell 2004;5:163–175.
Shukla SK, Purohit V, Mehla K et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017;32:71–87.
Huang C, Qiu Z, Wang L et al. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res 2012;72:655–665.
Li J, Akbani R, Zhao W et al. Explore, visualize, and analyze functional cancer proteomic data using the cancer proteome atlas. Cancer Res 2017;77:e51–e54.
Wei D, Kanai M, Jia Z, Le X, Xie K. Kruppel-like factor 4 induces p27Kip1 expression in and suppresses the growth and metastasis of human pancreatic cancer cells. Cancer Res 2008;68:4631–4639.
Yan Y, Li Z, Kong X et al. KLF4-mediated suppression of CD44 signaling negatively impacts pancreatic cancer stemness and metastasis. Cancer Res 2016;76:2419–2431.
Li L, Long J, Mise K et al. PGC1α is required for the renoprotective effect of lncRNA Tug1 in vivo and links Tug1 with urea cycle metabolites. Cell Rep 2021;36:109510.
Naveh-Many T, Silver J. Effects of calcitriol, 22-oxacalcitriol, and calcipotriol on serum calcium and parathyroid hormone gene expression. Endocrinology 1993;133:2724–2728.
Cheng J, Zhang W, Zhang X, Li X, Chen J. Efficacy and safety of paricalcitol therapy for chronic kidney disease: a meta-analysis. Clin J Am Soc Nephrol CJASN 2012;7:391–400.
Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med 2015;4:1033–1043.
Santana-Codina N, Roeth AA, Zhang Y et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 2018;9:4945.
Ying H, Kimmelman AC, Lyssiotis CA et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–670.
Rasmussen LS, Yilmaz MK, Falkmer UG et al. Pre-treatment serum vitamin D deficiency is associated with increased inflammatory biomarkers and short overall survival in patients with pancreatic cancer. Eur J Cancer (Oxford, England: 1990) 2021;144:72–80.
Fernández-Barral A, Costales-Carrera A, Buira SP et al. Vitamin D differentially regulates colon stem cells in patient-derived normal and tumor organoids. FEBS J 2020;287:53–72.
Hata T, Rajabi H, Takahashi H et al. MUC1-C activates the NuRD complex to drive dedifferentiation of triple-negative breast cancer cells. Cancer Res 2019;79:5711–5722.
Le Large TYS, El Hassouni B, Funel N et al. Proteomic analysis of gemcitabine-resistant pancreatic cancer cells reveals that microtubule-associated protein 2 upregulation associates with taxane treatment. Ther Adv Med Oncol 2019;11:1758835919841233.
Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resistance Updates Rev Commentaries Antimicrobial Anticancer Chemother 2015;23:55–68.
Ko AH, LoConte N, Tempero MA et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016;45:370–375.
Van Cutsem E, Tempero MA, Sigal D et al. Randomized phase III trial of pegvorhyaluronidase alfa with nab-paclitaxel plus gemcitabine for patients with hyaluronan-high metastatic pancreatic adenocarcinoma. J Clin Oncol Off J Am Soc Clin Oncol 2020;38:3185–3194.
Olive KP, Jacobetz MA, Davidson CJ et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (New York, NY) 2009;324:1457–1461.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–429.
Santiso-Mere D, Sone T, Hilliard GM, Pike JW, McDonnell DP. Positive regulation of the vitamin D receptor by its cognate ligand in heterologous expression systems. Mol Endocrinol (Baltimore, Md) 1993;7:833–839.
Miller CW, Morosetti R, Campbell MJ, Mendoza S, Koeffler HP. Integrity of the 1,25-dihydroxyvitamin D3 receptor in bone, lung, and other cancers. Mol Carcinogenes 1997;19:254–257.
Roy LD, Sahraei M, Subramani DB et al. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 2011;30:1449–1459.
Sahraei M, Bose M, Sanders JA et al. Repression of MUC1 promotes expansion and suppressive function of myeloid-derived suppressor cells in pancreatic and breast cancer murine models. Int J Mol Sci 2021;22:5587.
Tsutsumida H, Swanson BJ, Singh PK et al. RNA interference suppression of MUC1 reduces the growth rate and metastatic phenotype of human pancreatic cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res 2006;12:2976–2987.
Chaika NV, Gebregiworgis T, Lewallen ME et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA 2012;109:13787–13792.
Olou AA, King RJ, Yu F, Singh PK. MUC1 oncoprotein mitigates ER stress via CDA-mediated reprogramming of pyrimidine metabolism. Oncogene 2020;39:3381–3395.
Nagai K, Adachi T, Harada H, Eguchi S, Sugiyama H, Miyazaki Y. Dendritic cell-based immunotherapy pulsed with wilms tumor 1 peptide and mucin 1 as an adjuvant therapy for pancreatic ductal adenocarcinoma after curative resection: a phase i/iia clinical trial. Anticancer Res 2020;40:5765–5776.
Striefler JK, Riess H, Lohneis P et al. Mucin-1 protein is a prognostic marker for pancreatic ductal adenocarcinoma: results from the CONKO-001 study. Front Oncol 2021;11:670396.
Wang S, You L, Dai M, Zhao Y. Quantitative assessment of the diagnostic role of mucin family members in pancreatic cancer: a meta-analysis. Ann Transl Med 2021;9:192.
Acknowledgments
This study made use of the MD Anderson Cancer Center Functional Proteomics RPPA Core Facility, the Metabolomics Core Facility, and Research Animal Support Facility, supported by Cancer Center Support Grant CA016672. We thank Dawn Chalaire, scientific editor in the Research Medical Library at MD Anderson Cancer Center, for editing the manuscript. The authors thank Wei Li for the graphic arts used in the manuscript.
Funding
The work was supported by the National Cancer Institute (R01 CA198090 to R.S.B), the NIH-funded Texas Medical Center Digestive Disease Center Research Core Center Program P30DK056338 (to R.S.B and D.W), and grants from the Elsa U. Pardee Foundation, the University of Texas MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment, and the MD Anderson Cancer Center Institutional Research (to D.W).
Author information
Authors and Affiliations
Contributions
DW and RB conceived the study, designed the experiments, and wrote the manuscript. DW, LW, YL, XZ, and MH performed various portions of animal experiments and histologic analysis and the in vitro experiments. YL performed part of the bioinformatic data analysis. PY, LT, and PLL helped with IC-MS assay and data analysis. XZ provided conceptual feedback for the manuscript. All authors have read and agreed to the publication of the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no disclosures.
Ethical approval
The animal protocol and human subject protocol involved in this study were approved by the institutional Review Board of UT MD Anderson Cancer Center.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wei, D., Wang, L., Liu, Y. et al. Activation of Vitamin D/VDR Signaling Reverses Gemcitabine Resistance of Pancreatic Cancer Cells Through Inhibition of MUC1 Expression. Dig Dis Sci 68, 3043–3058 (2023). https://doi.org/10.1007/s10620-023-07931-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-023-07931-3