
Abstract
Background
The high mortality rate of decompensated cirrhosis underlines the need for new treatments. Experimental models of cirrhosis and its reported relationship with atherosclerotic cardiovascular disease have provided data supporting the rational use of statins in these patients. However, little is known about the safety of statins in this setting.
Aim
We evaluate the safety of chronic simvastatin treatment in patients with decompensated cirrhosis.
Methods
We conducted a prospective, open, uncontrolled, phase 2a trial in 30 patients with Child–Pugh class A (n = 6), B (n = 22), and C (n = 2) decompensated cirrhosis. The patients received standard treatment throughout the trial plus simvastatin 20 mg/day for 2 weeks and thereafter simvastatin 40 mg/day up to 1 year.
Results
Sixteen out of 30 patients (53.3%) showed adverse events, including gastrointestinal toxicity (36.7%), muscle injury (MI) (36.7%), and headache (13.3%). No liver injury was registered. Due to MI alone, simvastatin dosage was reduced in 23.4% of cases and transiently interrupted in 13.3%. Once these adverse events were overcome, simvastatin was resumed until the end of the trial. MI was associated with baseline MELD score > 12 (p = 0.035) and with baseline Child–Pugh class C. No MI was associated with final Child–Pugh score ≤ 6 (p = 0.030) or final Child–Pugh class A (p = 0.020).
Conclusions
Chronic treatment with simvastatin 40 mg/day in patients with decompensated cirrhosis was associated with several adverse events, being MI the only clinically significant one, which appears to be related to the simvastatin dosage and the degree of cirrhosis severity. Noticeably, no liver injury was recorded.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The high mortality rate of decompensated cirrhosis underlines the need for new treatments [1]. Experimental models of chronic liver disease have provided data supporting the rational use of statins due to their pleiotropic effects. It has been demonstrated that statins inhibit activation of hepatic stellate cells, reducing hepatic fibrogenesis. They also increase hepatic endothelial nitric oxide synthase levels, contributing to the improvement of endothelial dysfunction. Furthermore, they have chemopreventive effects, including induction of apoptosis and inhibition of angiogenesis in hepatocarcinoma [2].
Considering the reported relation between cirrhosis and cardiovascular atherosclerotic disease [3,4,5], statins could be considered as a therapeutic option [6]. Therefore, it was proposed that cirrhosis might be treated nonspecifically and in a cost-effective manner by using inexpensive, relatively safe, approved drugs such as statins [7].
However, little is known about the safety of statins in cirrhosis, with hepatotoxicity fears contributing to underutilization of statins by physicians in patients with abnormal aminotransferase values and/or viral hepatitis diagnosis [8]. Nevertheless, statins have been demonstrated to be safe in patients with chronic hepatic disease, even compensated cirrhosis [9]. For this reason, the Liver Expert Panel of the US National Lipid Association did not identify any scientific evidence to consider compensated cirrhosis as a Contraindication to the use of statins. On the contrary, the Liver Expert Panel believes that decompensated cirrhosis should not be considered an indication for treatments using statins, since these would not be a therapeutic option for patients with life-threatening illnesses [10]. The Bleeding Prevention With Simvastatin (BLEPS) study is the largest randomized controlled trial (RCT) of statins in patients with decompensated cirrhosis performed to date [11]. Six of the 69 patients (8%) treated with simvastatin 40 mg/day developed serious adverse events related to this drug. Furthermore, a systematic review and metaanalysis based upon observational databases over a large number of patients also showed that statins could reduce cirrhosis complications, hepatocarcinoma, and/or death in patients with chronic liver disease [12]. However, none of these studies evaluated the safety of statins as a primary endpoint.
This trial is designed to assess the safety of chronic simvastatin treatment in patients with decompensated cirrhosis.
Methods
Study Design and Place of Study
The present study is an investigator-initiated prospective, open, uncontrolled phase 2a trial. Patients were selected from the Hepatology Section of Dr. Carlos Bonorino Udaondo Hospital. The Ethics Committee for Clinical Research of Dr. Carlos Bonorino Udaondo Hospital approved the trial protocol. This trial was undertaken following the principles of the Helsinki Declaration, revised in Edinburgh in 2000. Written informed consent was obtained from all participants based on local Institutional Review Board requirements. The trial was conducted between July 2015 and December 2017.
Participants and Eligibility Criteria
The target population of the trial included patients with decompensated cirrhosis from outpatient clinical practice. The inclusion criteria were age over 18 years, either gender, and decompensated cirrhosis by ascites, variceal bleeding, hepatic encephalopathy, and/or jaundice. The exclusion criteria were Model for End-Stage Liver Disease (MELD) ≥ 20; use of recreational drugs in the 3 months previous to the trial start date; surgical portosystemic shunt or previous transjugular intrahepatic portosystemic shunt; thrombosis and/or portal vein cavernomatosis; hepatic encephalopathy grade ≥ II; hepatocellular carcinoma; no use of contraceptive methods by women of childbearing age; pregnant or lactating; having been indicated statin therapy or use of statins in the 3 months previous to the trial start date; hypersensitivity to statins; previous myopathy and/or stroke; liver transplant candidacy; human immunodeficiency virus infection; cancer; gastrointestinal or surgical diseases interfering with proper absorption of simvastatin; treatment with substrates and/or inhibitors of P-gp efflux transporter, cytochrome P4503A4, organic anion transporting polypeptides 1B1, and/or statin glucuronidation; or any other medical, psychiatric, or social condition that led the investigator to consider the patient inappropriate for the trial.
Interventions
Patients were included as participants of this trial at least 1 month after the last cirrhosis complication arose. This period of time was set for several reasons: to ensure that liver function was stable, with no new cirrhosis complications; to assess baseline biochemical data; to evaluate patients’ compliance with ambulatory follow-up; and to warrant simvastatin availability.
The following data were evaluated at first appointment: inclusion criteria, exclusion criteria, etiology of cirrhosis, previous cirrhosis complications, comorbidities, concomitant medication, physical examination, vital signs, biochemical values, pregnancy test when applicable, vitamin D supplementation for patients with serum levels < 30 IU/mL [13], Child–Pugh class and score, and MELD score.
The initial dosage of simvastatin was 20 mg/day (one tablet) for the first 2 weeks. In all patients, at day 15 of the study, the dosage of simvastatin was increased to 40 mg/day (two tablets), and this dosage was maintained for up to 1 year. In all cases, patients were advised to take simvastatin in the evening.
Monitoring visits were scheduled in the second week and then once a month until up to 1 year: no further follow-up was carried out after the trial. During these visits, the patients were subject to the following controls: physical examination, vital signs, laboratory, pregnancy test when applicable, compliance with simvastatin, Child–Pugh class and score, and MELD score evaluation. A checklist method was used [14] to assess frequent and potentially severe or serious adverse events such as gastrointestinal symptoms, headache, hepatitis symptoms, myalgia, and muscle weakness. The causal relationship between adverse events and simvastatin therapy was determined according to the researcher’s criteria. Simvastatin dosage reduction or transitory interruption was decided upon the occurrence of adverse events, while simvastatin discontinuation was decided upon the onset of serious adverse events. At the 6- and 12-month visits, additional abdominal ultrasound images were ordered for hepatocellular carcinoma screening and ascites detection.
Adherence to simvastatin was assessed on the basis of pill counts in dispensed boxes and was considered adequate with counts greater than 70%, regular with counts between 30% and 70%, and poor with counts less than 30%.
Endpoint
The endpoint is to assess simvastatin safety by considering the proportion of patients who developed adverse events and the proportion of patients with modified drug dosage due to the appearance of side effects, both related to chronic simvastatin therapy.
Adverse Events Associated with Simvastatin
Gastrointestinal Toxicity and Headache
The symptoms evaluated were constipation, diarrhea, abdominal pain, flatulence, and nausea [15]. Together with headache, these symptoms are the most common statin-associated adverse events reported in noncirrhotic patients [15].
Liver Injury
Liver injury was assessed according to the severity index recommended by Aithal et al. [16]. Liver injury severity was considered mild in patients presenting elevated alanine aminotransferase (ALT) ≥ fivefold above mean baseline value (MBV) and/or elevated alkaline phosphatase (ALP) ≥ twofold above MBV and bilirubin (BIL) concentration < twofold mean MBV; it was considered moderate in patients with elevated ALT and/or ALP concentrations similar to those for mild injury but with BIL concentration > twofold MBV or with symptomatic hepatitis; it was considered severe in patients with elevated ALT and/or ALP concentrations as mentioned above, BIL concentration > twofold MBV, and one of the following: international normalized ratio ≥ 1.5, ascites, hepatic encephalopathy, and/or other organ failure considered to be caused by liver injury.
Muscle Injury (MI)
MI was assessed according to the National Lipid Association Safety Expert Panel [17]. Statin-associated muscle adverse events include myalgia, myopathy, myositis, myonecrosis, and rhabdomyolysis. Myalgia is an unexplained muscle discomfort often described as “flu-like” symptoms with normal creatine kinase (CK) levels. Myopathy is determined by muscle weakness, which is not necessarily associated with elevated CK levels. A physical examination is needed to diagnose myositis. Myonecrosis condition is defined by the magnitude of serum CK elevation as mild when CK > threefold the upper limit of normal (ULN), moderate when CK ≥ tenfold ULN, and severe when CK ≥ 50-fold ULN. Clinical rhabdomyolysis is defined as myonecrosis plus myoglobinuria or renal failure (serum creatinine level increase ≥ 0.5 mg/dL). The ULN for CK used in the present work is 234 IU/L in females and 397 IU/L in males. Finally, the association between the severity of cirrhosis and the appearance—or not—of MI was evaluated through Child–Pugh class, Child–Pugh score, and MELD score, both at baseline and at study end.
Simvastatin Dosage Modifications
Simvastatin dosage modifications refer to adjustments that were necessary due to the appearance of simvastatin-associated adverse events, according to the following recommendations: (1) simvastatin dosage was reduced to 10 mg/day in the presence of mild liver injury, myalgia, or myopathy; (2) simvastatin administration was transiently interrupted in case of moderate liver injury, myositis, or myonecrosis; and (3) simvastatin administration was permanently interrupted in case of severe liver injury or clinical rhabdomyolysis. The above-mentioned adverse events, leading to changes in simvastatin dosage, were considered clinically significant.
Weekly clinical and biochemical controls were undertaken to monitor simvastatin-associated adverse event/s and to decide on additional adjustments of simvastatin dosage. These adjustments continued until the adverse events were overcome, evaluated through normalization of biochemical disorders and disappearance of symptoms. If simvastatin was transiently interrupted or reduced to 10 mg/day and the adverse event was reverted, simvastatin intake was resumed at 10 mg/day and then increased to the highest dosage tolerable for the patient.
Sample Size
The reported prevalence of adverse events associated with simvastatin in patients without liver disease is 9.0% [15] (null hypothesis). Our initial estimation of adverse events due to simvastatin in patients with decompensated cirrhosis was 30.0% (alternative hypothesis). The bilateral chi-square test, assuming a type I error of 5.0% (p value < 0.05) and a type II error of 0.10 (90% power), supported that the sample size should be at least 30 patients in the present study. The sample size calculation was performed using the G*Power version 3.1.9.4 sample calculation program from the University of Kiel, Germany.
Interim Analysis
Considering that the endpoint of the trial is simvastatin safety, an interim analysis was undertaken after patient number 10 finished the study. These results were compared with available safety data found in literature [15,16,17]. The Data Monitoring Committee formed at the beginning of the trial included a pharmacologist, a biostatistician, and a hepatologist, who had access to the interim analysis and recommended to continue the study. This decision was shared with the Ethics Committee for Clinical Research of Dr. Carlos Bonorino Udaondo Hospital.
Statistical Analysis
A Kolmogorov–Smirnov test was used to evaluate whether the quantitative variables had a normal distribution. Data are expressed as mean and standard deviation (SD), and median and interquartile range (IQR) for numerical variables, and as percentage for categorical ones. Means were compared using a Student’s t-test for dependent and independent samples. When a variable did not follow a normal distribution, a nonparametric Wilcoxon test was used. Qualitative data were tabulated using double-entry tables, and comparisons were made using a bilateral chi-square test with Yates correction or a Fisher exact test, or comparison of proportions, as appropriate. Receiver operating characteristic (ROC) curves were used to assess the diagnostic accuracy of the baseline MELD score value that best predicted MI, and the diagnostic accuracy of the end-of-trial Child–Pugh score value that best predicted no MI, to determine the cutoff values with the highest sensitivity and specificity. p-Value < 0.05 was considered statistically significant. The statistical analysis was performed using the SPSS 25.0 statistical package program (IBM).
Results
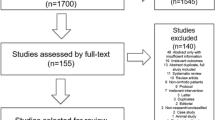
Sixty-five patients from outpatient clinical practice were screened between July 2015 and December 2016. Out of those 65 patients, 35 were excluded for different reasons, leaving 30 patients to be included in the trial (Fig. 1).

Flowchart showing disposition of patients
The baseline parameters are presented in Table 1. The median age was 56.6 years, and the majority of patients (67%) were male. The most frequent etiology of cirrhosis was alcohol, but only 13% of these patients were actively drinking when they were included in the trial. Their past and current history of cirrhosis complications are presented in Table 1. Ascites was the most frequent complication. Before their inclusion in this trial, 83% of patients were diagnosed with ascites, but at inclusion in this trial, this percentage had decreased to 63%. Some patients had more than one cirrhosis complication. In general, liver function was moderately impaired, with a Child–Pugh score of 7.3 ± 1.3 and only two patients being Child–Pugh class C, while the MELD score was 12.3 ± 3.3. One interesting finding in the biochemical parameters was hypovitaminosis D, which was present in 90% of the patients.
Characteristics of Simvastatin Treatment
Patients remained on simvastatin for a median of 52 weeks (range 46–52 weeks). Adherence to treatment was considered adequate in 100% of patients. The median follow-up time was 359 days (IQR 355–365 days).
Adverse Events Associated with Simvastatin
Percentages of total adverse events, each adverse event, and clinically significant adverse events are shown in Fig. 2. The proportion of patients with adverse events associated with simvastatin was greater (53.3%) than the proportion of patients with adverse events expected in this trial (30%), (p = 0.12).

Summary of simvastatin-related adverse events in study group (n = 30). Clinically significant adverse events: adverse events that led to changes in simvastatin dosage
Gastrointestinal Toxicity (GIT) and Headache
Adverse symptoms related to simvastatin were flatulence (23%), constipation (17%), abdominal pain (7%), diarrhea (3.5%), and nausea (3%). Some patients referred to more than one symptom. Nevertheless, no patient had their simvastatin dosage modified, neither reduced, transiently interrupted, nor permanently discontinued, due to GIT and/or headache (13.3%).
Liver Injury
No patient developed liver injury. Furthermore, comparing values at the end of the trial versus baseline, serum aspartate aminotransferase (AST) slightly decreased, serum ALT remarkably decreased (32 ± 16 versus 39 ± 20 IU/L, respectively; p = 0.09), and serum ALP decreased significantly (119 ± 48 versus 147 ± 67 IU/L, respectively; p = 0.02).
Muscle Injury
Out of 30 patients, 11 developed MI (36.7%). As presented in Table 2, 7 out of 30 patients developed myalgia (23.4%), and 4 out of 30 developed myonecrosis (13.3%). However, rhabdomyolysis was not detected in any patient. All patients had reached simvastatin 40 mg/day before MI started. In all patients, MI was related to simvastatin. Nine out of 11 patients with MI (81.8%) reported muscle aches, and 2 out of 4 patients with myonecrosis did not notify any muscle symptom. No patient reported other kind of muscle pain. As also presented in Table 2, muscle aches were symmetrically located in 73% of patients. Besides, 73% of patients (not necessarily the same patients) with MI developed muscle aches and/or serum CK concentration increases within the first 12 weeks after starting simvastatin therapy. Furthermore, serum AST and ALT concentrations were within normal values in the myalgia group and increased in the myonecrosis group (314 ± 157 and 144 ± 107 IU/L, respectively).
Note that all patients with myalgia had simvastatin dosage reduced to 10 mg/day (23.4%) and that all patients with myonecrosis had simvastatin transiently interrupted (13.3%). Because of these simvastatin dosage modifications, muscle pain was relieved and/or CK serum concentration normalized within 2–7 weeks. After MI was overcome, all patients continued or restarted simvastatin intake at 10 mg/day and completed the study with this dosage, or with the dosage that they were able to tolerate up to 40 mg/day (Table 3). Finally, no patients developed rechallenge symptoms and/or serum CK concentration increases.
Furthermore, the association between MI or no MI and cirrhosis severity was analyzed through Child–Pugh class, Child–Pugh score, and MELD score. A significantly greater baseline MELD score was observed within the group with MI compared with the group without MI (p = 0.035). Moreover, the only two Child–Pugh class C patients included in the study developed myonecrosis, and the other two patients with myonecrosis were Child–Pugh class B. When comparing the group without MI versus the group with MI, a significant improvement in Child–Pugh class (p = 0.020) and a lower value of Child–Pugh score (p = 0.030) were found at the end of the trial. These results are outlined in Table 4. However, ROC analysis revealed a cutoff value > 12 for baseline MELD score to differentiate patients with MI from patients without MI, with sensitivity of 72.7%, specificity of 73.7%, and area under the ROC curve of 0.73; and a cutoff value ≤ 6 for end-of-trial Child–Pugh score to differentiate patients without MI from patients with MI, with sensitivity of 63.2%, specificity of 72.7%, and area under the ROC curve of 0.71. Finally, note that no significant differences were observed between patients with or without MI concerning alcohol as etiology of cirrhosis, administration of diuretics, or baseline serum vitamin D concentrations.
Simvastatin Dosage Modifications
As presented in Table 3, simvastatin dosage was reduced or transiently interrupted due to MI; for this reason, it was considered to be the only clinically significant adverse event, as shown in Fig. 2. No patient required permanent discontinuation of simvastatin for severe liver injury, clinical rhabdomyolysis, or any other serious adverse event.
Discussion
In this prospective, open, uncontrolled phase 2a safety study, chronic simvastatin treatment in patients with decompensated cirrhosis resulted in the appearance of adverse events in 53.3% of patients. The side effects recorded in this trial were gastrointestinal toxicity (36.7%), headache (13.3%), and MI (36.7%), although no liver injury was registered. Despite the high proportion of cirrhotic patients with the above-mentioned side effects, none of these effects prevented chronic treatment with simvastatin. Nevertheless, due to MI, simvastatin dosage was reduced in 23.4% of patients and transiently interrupted in 13.3% of patients. Finally, MI appeared to be related to the dosage of simvastatin and severity of cirrhosis, and no patient had simvastatin permanently discontinued due to serious adverse events.
The most concerning adverse event of statins is MI [18]. In the present trial, the proportion of MI was 36.7%, which is higher than that observed in subjects without chronic liver disease, ranging from 1% to 5% in RCTs and from 11% to 29% in observational studies [19]. Besides, the BLEPS trial showed that 2.8% of patients who received simvastatin 40 mg/day developed rhabdomyolysis, and they had advanced liver disease (bilirubin > 5 mg/dL) [11]. As this outcome is greater than that observed in the general population (0.009–0.1%) [19], Abraldes et al. argue that patients with severe hepatic impairment can develop muscle damage at a lower dosage of statins than the general population [11]. Simvastatin is mainly metabolized by hepatic CYP3A4, known as the first-pass effect. However, we were not aware of simvastatin pharmacokinetic studies in patients with cirrhosis. In this context, Albarmawi et al. evaluated the pharmacokinetics of midazolam—also principally metabolized by liver CYP3A4—in patients with cirrhosis and found a good correlation between Child–Pugh class and MELD score (A/≤ 9, B/10–14, and C/≥ 15). Likewise, they found that both scores correlated well and inversely with midazolam clearance. Reduction of the hepatic first-pass effect of drugs such as midazolam and simvastatin could be related to high plasma concentrations and increased risk of adverse events [20]. Therefore, these findings can help explain the possible association between the baseline severity of cirrhosis (MELD score > 12 and Child–Pugh class C) and the development of MI. Finally, regarding the impact of these outcomes upon clinical care and the safety of simvastatin administration in patients with decompensated cirrhosis, it is suggested that MELD score and Child–Pugh class should be evaluated before administering simvastatin at 40 mg/day dosage.
In this regard, the LIVERHOPE-SAFETY trial was recently published [21]. This is the first RCT to analyze the safety of two different dosages of simvastatin (20 mg/day, 40 mg/day plus rifaximin, or placebo of both drugs during 3 months) in patients with decompensated cirrhosis. Pose et al. found that the simvastatin 40 mg/day group, in comparison with the other groups, was associated with a significant increase in liver and muscle toxicities requiring treatment withdrawal. They concluded that simvastatin dosage should be 20 mg/day in future studies in patients with decompensated cirrhosis. However, note that, in the current trial, no patient showed liver injury even after 12 months of simvastatin administration, and liver enzymes improved at the end of the study as compared with baseline levels, as already described by other authors in patients on statin therapy with and without chronic liver disease [9, 22]. Moreover, there was a different simvastatin dosage management approach in patients with MI when comparing both trials. Nevertheless, this trial showed that the final dosage of simvastatin was 10 or 20 mg/day in most patients with MI. Consequently, we subscribe to the LIVERHOPE-SAFETY trial conclusions, in that the dosage of simvastatin must be lower than 40 mg/day for patients with decompensated cirrhosis.
However, when comparing patients without MI versus those with MI at the end of the study versus baseline, a noticeable improvement was found in Child–Pugh score and Child–Pugh class. These results would suggest a reduction of cirrhosis severity at the end of the study. Due to the characteristics of the trial, these findings are merely empirical. In experimental models, it was proved that statins act beneficially through various pleiotropic mechanisms to improve chronic liver diseases [2]. Figure 3 shows that nine patients with baseline Child–Pugh class B became Child–Pugh class A at the end of the trial.

Association between patients without muscle injury at baseline and end of trial and cirrhosis severity according to Child–Pugh class A and B. Note the change of set for nine patients from Child–Pugh class B group at baseline to Child–Pugh class A group at end of trial
The strength of this trial is that, with a low budget and in a relatively short term, it was shown that the adverse events in patients with decompensated cirrhosis are frequent (gastrointestinal symptoms) and could be severe or serious (MI) due to chronic simvastatin therapy [23]. A possible limitation to this trial is the relatively small sample size: regarding this, it was explicitly calculated to investigate the safety of simvastatin therapy in patients with decompensated cirrhosis, and it was based on the only published safety data available (until 2015, for patients without liver disease) [15]. However, in 2016, with this trial already started, Abraldes et al. published that 8% of patients with decompensated cirrhosis who received simvastatin showed adverse events related to the study drug [11] and that the percentage of patients with significant clinical adverse events reached 36.7%. Therefore, it was assumed that the number of patients included in this trial should be considered sufficient. Moreover, previous studies have demonstrated that the checklist method identified more adverse events than other data-collecting procedures [14]. However, when the symptoms present a high background rate, such as gastrointestinal and muscle symptoms, the checklist method tends to capture a large number of adverse events of doubtful clinical importance [23]. Therefore, in two placebo-controlled simvastatin RCTs collecting muscle symptoms through a checklist, 33–56% of participants taking placebo or simvastatin had adverse events [24, 25]. One of the design drawbacks of this study is the lack of a control group as well as not analyzing the potential beneficial effects that alcohol abstinence may have had, considering that alcohol consumption was the primary etiology of cirrhosis in this trial since only 13% of patients continued drinking during the study [26].
In conclusion, this trial showed that chronic simvastatin treatment in patients with decompensated cirrhosis up to 1 year was associated with a high frequency of adverse events, although no liver injury was registered. Moreover, simvastatin dosage modification was only necessary to alleviate MI, which in turn appears to be related to the simvastatin dosage and the severity of cirrhosis. Consequently, a simvastatin dosage of 40 mg/day should not be prescribed in patients with cirrhosis MELD score > 12 because of the high rate of muscle adverse events or in Child–Pugh class C patients due to potential severe MI. Considering the design of this trial, these conclusions require additional validation by RCTs. The new trials should also assess the safety and efficacy of a lower dosage of simvastatin for Child–Pugh class A and B patients and those with MELD score < 12, the simvastatin dosage management approach in case of adverse events, and simvastatin benefits in patients with decompensated cirrhosis—with or without current alcohol consumption. Finally, it would be advantageous to study the pharmacokinetics of simvastatin in cirrhotic patients.
Abbreviations
- RCT:
-
Randomized controlled trial
- MELD:
-
Model for End-Stage Liver Disease
- ALT:
-
Alanine aminotransferase
- MBV:
-
Mean baseline values
- ALP:
-
Alkaline phosphatase
- BIL:
-
Bilirubin
- MI:
-
Muscle injury
- CK:
-
Creatine kinase
- ULN:
-
Upper limit of normal
- IQR:
-
Interquartile range
- ROC:
-
Receiver operating characteristic
- GIT:
-
Gastrointestinal toxicity
- AST:
-
Aspartate aminotransferase
References
D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-231.
Schierwagen R, Uschner FE, Magdaleno F, et al. Rationale for the use of statins in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;312:G407-G412.
Kalaitzakis E, Rosengren A, Skommevik T, et al. Coronary artery disease in patients with liver cirrhosis. Dig Dis Sci. 2010;55:467-475. https://doi.org/10.1007/s10620-009-0738-z
Wehmeyer MH, Heuer AJ, Benten D, et al. High rate of cardiac abnormalities in a postmortem analysis of patients suffering from liver cirrhosis. J Clin Gastroenterol. 2015;49:866-872.
Danielsen KV, Wiese S, Hove J, et al. Pronounced coronary arteriosclerosis in cirrhosis: Influence on cardiac function and survival? Dig Dis Sci. 2018;63:1355-1362. https://doi.org/10.1007/s10620-018-5006-7
Stone NS, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
Tsochatzis EA, Bosch J, Burroughs AK. New therapeutic paradigm for patients with cirrhosis. Hepatology. 2012;56:1983-1992.
Rzouq FS, Volk ML, Hatoum HH, et al. Hepatotoxicity fears contribute to underutilization of statin medications by primary care physicians. Am J Med Sci. 2010;340:89-93.
Lewis JH, Mortensen ME, Zweig S, et al. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453-1463.
Cohen DE, Anania FA, Chalasani N. An assessment of statin safety by hepatologists. Am J Cardiol. 2006;97:77C-81C.
Abraldes J, Villanueva C, Aracil C, et al. Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology. 2016;150:1160-1170.e3.
Kamal S, Khan M, Seth A, et al. Beneficial effects of statins on the rates of hepatic fibrosis, hepatic decompensation, and mortality in chronic liver disease: A systematic review and meta-analysis. Am J Gastroenterol. 2017;112:1495-505.
Lee P, Greenfield JR, Campbell LV. Vitamin D insufficiency-a novel mechanism of statin-induced myalgia? Clin Endocrinol. 2009;71:154-155.
Bent S, Padula A, Avins AL. Brief communication: Better ways to question patients about adverse medical events. A randomized, controlled trial. Ann Intern Med. 2006;144:257–261.
Boccuzzi S, Bocanegra T, Walker F, et al. Long-term safety and efficacy of simvastatin. Am J Cardiol. 1991;68:1127-1131.
Aithal G, Watkins P, Andrade R, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89:806-815.
Rosenson R, Baker S, Jacobson T, et al. An assessment by the statin muscle safety task force: 2014 update. J Clin Lipidol. 2014;8:S58-S71.
Simic I, Reiner Z. Adverse effects of statins - myths and reality. Curr Pharm Des. 2015;21:1220-1226.
Thompson P, Panza G, Zaleski A, et al. Statin-associated side effects. J Am Coll Cardiol. 2016;67:2395-2410.
Albarmawi A, Czock D, Gauss A, et al. CYP3A activity in severe liver cirrhosis correlates with Child-Pugh and model for end-stage liver disease (MELD) scores. Br J Clin Pharmacol. 2014;77:160-169.
Pose E, Napoleone L, Amin A, et al. Safety of two different doses of simvastatin plus rifaximin in decompensated cirrhosis (LIVERHOPE-SAFETY): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Gastroenterol Hepatol. 2020;5:31-41.
Tikkanen MJ, Fayyad R, Faergeman O, et al. Effect of intensive lipid lowering with atorvastatin on cardiovascular outcomes in coronary heart disease patients with mild-to-moderate baseline elevations in alanine aminotransferase levels. Int J Cardiol. 2013;168:3846-3852.
Newman CB, Preiss D, Tobert JA, et al. Statin safety and associated adverse events: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2019;39:e38-e81.
Keech A, Collins R, MacMahon S, et al. Three-year follow-up of the Oxford Cholesterol Study: assessment of the efficacy and safety of simvastatin in preparation for a large mortality study. Eur Heart J. 1994;15:255-269.
Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7-22.
Verrill C, Markham H, Templeton A, et al. Alcohol-related cirrhosis-early abstinence is a key factor in prognosis, even in the most severe cases. Addiction. 2009;104:768-774.
Acknowledgments
We thank Stella Hirmas, Lab Technician, for her collaboration; Mrs. María Eugenia Muñoz and Lic. Harry Rodger for providing technical language advisory; and Laboratorio Roemmers, Buenos Aires, Argentina for supplying simvastatin tablets.
Funding
No funding was received to design or undertake this study or to write this manuscript. When studies are initiated and conducted in Argentina by the researchers, there is no funding from any public or private institution as they are made with financial contributions from the researchers. Granted scholarships: Florencia Pollarsky was granted the “Beca Estímulo Florencio Fiorini para Investigación en Medicina año 2016” for the project identified as "Study of safety, survival, and quality of life in patients with decompensated cirrhosis receiving conventional treatment plus simvastatin.” Trial registration number: Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), Dirección de Evaluación y Registro de Medicamentos, INAME, Comisión de Ensayos Clínicos. File no. 1-47-17211-14-5, 20 February 2015.
Author information
Authors and Affiliations
Contributions
Guarantor of the article: A.E.M. Acquisition of data: A.E.M., F.P., M.M., M.C., C.M., D.A., and G.R. Analysis and interpretation of data: A.E.M., F.P., P.S., H.V., and G.R. Drafting of the manuscript: A.E.M., F.P., P.S., H.V., and G.R. Critical review of the manuscript for relevant intellectual content: A.E.M., F.P., M.M., M.C., C.M., D.A., P.S., H.V., and G.R. Statistical analysis: P.S. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Muñoz, A.E., Pollarsky, F., Marino, M. et al. Safety of Chronic Simvastatin Treatment in Patients with Decompensated Cirrhosis: Many Adverse Events but No Liver Injury. Dig Dis Sci 66, 3199–3208 (2021). https://doi.org/10.1007/s10620-020-06630-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-020-06630-7