
Abstract
Background and Aims
In the present study, we investigated the differentially expressed genes (DEGs), pathways and immune cell infiltration characteristics of pediatric and adult ulcerative colitis (UC).
Methods
We conducted DEG analysis using the microarray dataset GSE87473 containing 19 pediatric and 87 adult UC samples downloaded from the Gene Expression Omnibus. Gene ontology and pathway enrichment analyses were conducted using Metascape. We constructed the protein–protein interaction (PPI) network and the drug–target interaction network of DEGs and identified hub modules and genes using Cytoscape and analyzed immune cell infiltration in pediatric and adult UC using CIBERSORT.
Results
In total, 1700 DEGs were screened from the dataset. These genes were enriched mainly in inter-cellular items relating to cell junctions, cell adhesion, actin cytoskeleton and transmembrane receptor signaling pathways and intra-cellular items relating to the splicing, metabolism and localization of RNA. CDC42, POLR2A, RAC1, PIK3R1, MAPK1 and SRC were identified as hub DEGs. Immune cell infiltration analysis revealed higher proportions of naive B cells, resting memory T helper cells, regulatory T cells, monocytes, M0 macrophages and activated mast cells in pediatric UC, along with lower proportions of memory B cells, follicular helper T cells, γδ T cells, M2 macrophages, and activated dendritic cells.
Conclusions
Our study suggested that hub genes CDC42, POLR2A, RAC1, PIK3R1, MAPK1 and SRC and immune cells including B cells, T cells, monocytes, macrophages and mast cells play vital roles in the pathological differences between pediatric and adult UC and may serve as potential biomarkers in the diagnosis and treatment of UC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple clinical observations as well as epidemiologic studies show the differences in the extent and severity of disease between pediatric-onset ulcerative colitis (UC) and its adult counterpart. Most studies on UC have demonstrated that up to 60–70% of pediatric-onset UC presents with a "panenteric" phenotype compared to 20–30% of adult-onset UC patients [1,2,3]. Within 10 years of diagnosis, more than 40% of pediatric-onset UC patients had undergone colectomy compared to less than 20% in adult-onset UC patients [2]. In addition to having a more severe phenotype with rapid progression of inflammatory disorders of the gastrointestinal tract, pediatric-onset UC patients are at higher risk of developing extra-intestinal manifestations (EIM) than adult-onset UC patients, which is predictive of more a severe disease outcome [4]. Cortisol is widely used for the treatment of adult-onset UC; however, cortisol is not recommended for use in pediatric-onset UC patients because of the high rate of refractory disease and drug dependence and detrimental effects on growth and bone density accrual [5, 6]. According to the first pediatric specific UC guidelines jointly produced by ECCO and ESPGHAN, indefinite maintenance therapy is necessary for the treatment of adult-onset UC patients, which combines high doses of 5-ASA with thiopurines or the anti-TNF biologic infliximab [7, 8]. Although several studies have focused on the differences in clinical characteristics and administration strategies between pediatric and adult UC, transcriptomic comparisons at the molecular level are few reported and the differences in gene expression, biological processes and pathways between pediatric and adult UC are still unknown [9,10,11].
The pathological tissue damage in UC is partly driven by exaggerated and inappropriate immune responses involving both the innate and adaptive immune systems. Activation or inactivation of various types of immune cells in UC contributes to disease initiation, augmentation, and perpetuation by regulating the suppression, maintenance or promotion of immune responses [12,13,14]. However, to our knowledge, no studies focusing on a comparison between the characteristics of immune cell infiltration in pediatric and adult UC have yet been reported.
A comparison of the gene expression profile between two subtypes of a disease is useful to identify reliable biomarkers and novel treatment targets for the improvement of therapeutic efficacy and personalized therapy [15,16,17,18,19,20]. Bioinformatics analysis of gene expression profiles is a promising tool for the comprehensive identification of the differentially expressed genes (DEGs) and biological pathways involved in the development of UC in pediatric and adult patients at the whole genome level. In our current study, gene microarray dataset GSE87473 was obtained from Gene Expression Omnibus (GEO), which contains 19 pediatric and 87 adult UC samples. We conducted differential gene expression analysis, followed by Gene Ontology (GO) and pathway enrichment analysis of DEGs. The protein–protein interaction (PPI) network of DEGs was then constructed and key modules were mined and functionally annotated. Furthermore, we investigated and compared the immune cell infiltration characteristics between pediatric and adult UC using a novel gene-based approach CIBERSORT to deconvolute gene expression data and determine 22 immune cell types in UC samples [21].
Materials and Methods
Microarray Data
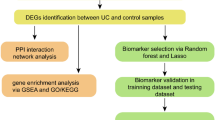
The procedures used in our study are illustrated in the flow chart shown in Fig. 1. The mRNA microarray dataset GSE87473 deposited by Li et al. was downloaded from Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/). The inclusion criteria for the gene expression profile are set as follows: (1) The samples detected are colon tissues from homo sapiens, (2) tissues are diagnosed with pediatric and adult UC tissues, (3) gene expression profiling of mRNA, (4) the number of pediatric or adult UC samples was more than 15. The database contained colon biopsies from 19 pediatric and 87 adult UC patients, and the GPL13158 [HT_HG-U133_Plus_PM] Affymetrix HT HG-U133+PM Array Plate was utilized to obtain gene expression profiles. As shown in Figure S1, the box plot of the distribution of value data is median-centered, indicating that the data are cross-comparable and appropriate for further investigations.

Flowchart of the study procedures
Identification of Differentially Expressed Genes
The DEGs between pediatric and adult UC colon samples were screened using the interactive web tool GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/), which is capable of comparing and analyzing two sets of GEO array data [22]. P values < 0.05 and |log FC|> 2.5 were set as cut-off standards and considered to indicate statistical significance. The volcano plot of DEGs from GSE87473 were constructed using ImageGP (https://www.ehbio.com/ImageGP/index.php/Home/Index/Volcanoplot.html).
Gene Ontology (GO) and Pathway Enrichment Analysis
Gene Ontology (GO) analysis is a major tool for annotating genes, gene products and sequences. The Kyoto Encyclopedia of Genes and Genomes (KEGG) is an integrated database resource for biological interpretation of genome sequences and other high-throughput data to identify functional and metabolic pathways. Metascape (https://metascape.org) is an online program for gene functional enrichment analysis by analyzing gene/protein lists and for intuitive and visually representation of the results [23]. We used the "Custom Analysis" mode of Metascape to perform GO [including Biological process (BP), cellular component (CC) and molecular function (MF)] and KEGG pathway analysis. P < 0.01 was considered to indicate significantly enrichment of the DEGs.
PPI Networks and Hub Gene Definition
The PPI networks of DEGs were generated using the Search Tool for the Retrieval of Interacting Genes (STRING 11.0; https://string.embl.de/) database, with a confidence score > 0.9 (highest confidence) set as the cut-off criterion [24]. The PPI network was then visualized using Cytoscape 3.6.1 software and hub modules of the network were screened using the Molecular Complex Detection (MCODE) plug-in with MCODE scores > 10, with the degree of connectivity cut-off, node score cut-off, k-core, and maximum depth set as 2, 0.2, 2 and 100, respectively [25, 26]. In addition, using the NetworkAnalyzer, hub genes with a degree > 60 in the PPI network were defined as hub genes [27].
Analysis of Drug–Target Interactions
The drug–gene interaction database (DGIdb, v3.0.2, https://www.dgidb.org/) was applied to predict the drug–gene interactions using the list of hub DEGs with the filter condition ‘FDA Approved’ [28]. Drug–gene interaction was constructed and visualized by Cytoscape.
Immune cell Infiltration Evaluation and Analysis
CIBERSORT is an analytical tool used for sensitive and specific quantification of the relative levels of 22 phenotypes of human hematopoietic cells within a complex gene expression mixture (https://cibersort.stanford.edu) [21]. The CIBERSORT P value reflects the statistical significance of the deconvolution results across all cell subsets, which is used to filter out deconvolution with less significant fitting accuracy. Normalized gene expression profiles from 87 adult UC and 19 pediatric UC patients were uploaded to the CIBERSORT web portal (https://cibersort.stanford.edu), and the number of permutations was set to 500. Immune cell profiles of samples that met the CIBERSORT P < 0.05 requirements were then obtained.
Statistical Analysis
Two-tailed Student's t-test was used to analyze the differences between immune cell fractions of eligible adult and pediatric UC samples using GraphPad Prism 7.0 software (GraphPad Software, Inc.). Correlation analysis of immune cell proportions in adult and pediatric UC samples was performed using the OmicShare tools, which is a free online platform for data analysis (https://www.omicshare.com/tools). Correlation matrixes of adult and pediatric UC were then constructed.
Results
Identification of DEGs
A total of 19,779 genes were detected in colon biopsies from 19 pediatric and 87 adult UC patients, of which 1700 [694 (40.8%) upregulated and 1006 (59.2%) downregulated] were identified as DEGs in the pediatric UC samples compared to adult UC samples (Fig. 2). The most significantly upregulated and downregulated genes were folate receptor 1 (FOLR1) (logFC = 7.2786365) and transcription factor 4 (TCF4) (logFC = − 4.5852488), respectively (Table S1).

Volcano plot of DEGs selection. Red dots represent upregulated genes and green dots represent downregulated genes
Enrichment Analysis of DEGs
GO and KEGG pathway enrichment analyses of overlapping DEGs showed a high level of enrichment in numerous functional groups and pathways. The 694 upregulated DEGs were mainly enriched in ‘anchoring junction’ (GO: 0070161), ‘cell junction organization’ (GO: 0034330), ‘regulation of actin cytoskeleton’ (hsa04810), ‘supramolecular fiber organization’ (GO: 0097435), ‘chromatin’ (GO: 0000785), and ‘cell adhesion molecule binding’ (GO: 0050839) (Fig. 3a, b). The 1006 commonly downregulated DEGs were mainly enriched in ‘regulation of mRNA metabolic process’ (GO: 1903311), ‘RNA localization’ (GO: 0006403), ‘ribonucleoprotein complex biogenesis’ (GO: 0022613), ‘RNA splicing via transesterification reactions’ (GO: 0000375), ‘regulation of cell morphogenesis’ (GO: 0022604), and ‘regulation of response to endoplasmic reticulum’ (GO: 1905897) (Fig. 3c, d).

Functional enrichment analysis of DEGs. a, c Top 20 clusters from Metascape pathway enrichment analysis of up- and downregulated DEGs colored according to P values. b, d Network of the top 20 enriched terms colored according to cluster ID. Nodes of the same color are typically close to each other
PPI Network Analysis and Hub Gene Identification
The PPI network of DEGs was constructed and visualized using Cytoscape based on the STRING output with highest confidence and consisted of 893 nodes (genes) and 5143 edges (interactions) (Fig. 4). After using the MCODE plug-in to identify modules from the PPI network, the top three central modules with MCODE scores > 10 were selected. Module 1 with MCODE scores of 32.431 consisted of 66 nodes and 1054 edges. Module 2 with MCODE scores of 17.417 consisted of 49 nodes and 418 edges. Module 3 with MCODE scores of 13.111 consisted of 46 nodes and 295 edges (Fig. 5) (Table 1). Through analysis of GO-BP and KEGG pathway enrichment, Module 1 was found to be mainly enriched in ‘RNA splicing via transesterification reactions with bulged adenosine as the nucleophile’ (GO: 0000377) and ‘spliceosome’ (hsa03040). Module 2 was mainly enriched in ‘cell division’ (GO: 0051301) and ‘organelle localization’ (GO: 0051640), and Module 3 was mainly enriched in ‘regulation of megakaryocyte differentiation’ (GO: 0045652) and ‘post-translational protein modification’ (GO: 0043687) (Table 2).

The protein–protein interaction network of the common nodes. Up- and downregulated DEGs are marked in red and green, respectively, and the size of the node represents the degree of connectivity

Modules that contain hub genes of the PPI network. Up- and downregulated DEGs are marked in red and green, respectively
DEGs with a high degree of centrality (> 60) were then identified as hub nodes in this PPI network. These hub nodes were identified as cell division control protein 42 (CDC42; degree = 97), DNA-directed RNA polymerase II subunit RPB1 (POLR2A; degree = 84), Ras-related C3 botulinum toxin substrate 1 (RAC1; degree = 84), phosphatidylinositol 3-kinase regulatory subunit alpha (PIK3R1; degree = 71), mitogen-activated protein kinase 1 (MAPK1; degree = 69) and proto-oncogene tyrosine-protein kinase Src (SRC; degree = 66) (Table 3). Specifically, CDC42 was in Module 2, POLR2A was in Module 1, and MAPK1 and SRC were in Module 3. Besides, Module 8 that contains RAC1 and PIK3R1 and its GO-BP and KEGG pathway enrichment were also provided in Fig. 5, Tables 1 and 2.
Analysis of Drug–Target Interactions
Study of drug–target interaction is great important for drug discovery and design. Based on the hub DEGs in the PPI network, the drug–target interactions were predicted by the DGIdb, which provided a theoretical therapeutic mechanism of UC for reference (Fig. 6). Four hub DEGs including SRC, MAPK1, RAC1 and CDC42 were found theoretically important in the UC therapy and they were targeted by 15, 6, 2 and 1 FDA-approved drugs, respectively.

The drug–target interaction network of DEGs. Circle represents DEGs and square represents drug
Immune Cell Infiltration Characterization
The 22 immune cell types infiltrating all pediatric and adult UC samples were investigated according to the CIBERSORT algorithm. Nine pediatric and 75 adult UC samples that matched the requirements of CIBERSORT P < 0.05 were filtered out (Table S2). As a result, 22 immune cell types were detected in adult UC samples, while 20 immune cell types were detected in pediatric UC samples in addition to naive CD4 T cells and eosinophils. The immunocyte composition of the entire cohort is summarized in Fig. 7a and the subpopulations of immune cells identified by unsupervised hierarchical clustering are shown in Fig. 7b. Correlation analysis of pediatric and adult UC samples furtherly revealed a low to moderate correlation between various immunocyte subpopulation fractions (Fig. 7c, d). In addition, relative to adult UC samples, higher proportions of naive B cells, resting memory T helper cells, regulatory (Tregs) T cells, monocytes, and M0 macrophages as well as activated mast cells were detected in pediatric UC samples, along with lower proportions of memory B cells, follicular helper T cells, γδ T cells, M2 macrophages, and activated dendritic cells (Fig. 7e).

Immune infiltrate landscape of pediatric and adult ulcerative colitis samples. a Stacked bar chart representing deviations in immune infiltration in each sample. b Hierarchical clustering of adult and pediatric UC samples based on immune cell proportions. Red, white and blue indicate high, moderate and low proportions of immune cells, respectively. c, d Correlation matrix of the immunocyte proportions in pediatric (c) and adult (d) UC samples. Red and blue color represent positive and negative correlations, respectively. e Differences in proportions of each immune cell type in pediatric and adult UC samples. *P < 0.05 **P < 0.01 ***P < 0.001 ****P < 0.0001
Discussion
Pediatric-onset UC is more severe and extensive with a devastating prognosis compared with its adult counterpart; however, the genetic differences and characteristics of these two forms of the disease are still unknown. In the present study, we used genomic analysis to screen a total of 1700 DEGs from GSE87473 microarray dataset in the GEO. As was suggested by the GO and KEGG enrichment analysis, 694 upregulated DEGs were mainly manifested in inter-cellular items that relate to cell junctions, cell adhesion, actin cytoskeleton and transmembrane receptor signaling pathways. The 1006 downregulated DEGs were mainly manifested in intra-cellular items that relate to the splicing, metabolism and localization of RNA.
According to the PPI network of DEGs, six hub genes with high degrees of connectivity were identified: CDC42, POLR2A, RAC1, PIK3R1, MAPK1 and SRC. MAPK1 (also known as ERK2) is a member of the MAPK family, playing a major role in MAPK/ERK signaling, which promotes inflammatory responses and inflammatory cytokine production. The expression of (p)-ERK1/2 was increased in DSS-induced UC rats, and its inhibition reduced inflammation and promoted restoration of epithelial barrier function [29,30,31,32,33,34,35]. CDC42 and RAC1 are small GTPases of the Rho family that control the switch between the active GTPbound and inactive GDP-bound forms. These molecules have similar biological characteristics that stabilize the intestinal epithelial barrier by promoting various cellular processes of intestinal epithelial cells (IECs) such as proliferation, adhesion, motility and actin cytoskeletal organization [36,37,38]. In UC, high expression of miR-15a stimulated by TNFα was found to directly target CDC42, causing low levels of expression compared to that in the healthy group [39]. Downregulated CDC42 in UC leads to decreased expression of zona occludens (ZO)-1 and E-cadherin, which negatively regulate intestinal epithelial junctions and disrupt the epithelial barrier [39]. Through activating the NADPH oxidase Nox1, Rac1 promotes the generation of reactive oxygen species (ROS) and recruitment of neutrophils in injured intestinal tissues, which promote intestinal epithelial wound repair by positively regulating adhesion of IECs to the matrix [40]. However, persistent neutrophil infiltration and the release of proinflammatory cytokines induced by Rac1 were found to cause destruction of the intestinal epithelium, which contributes to the development of UC [41,42,43]. In addition, two single nucleotide polymorphisms (SNP) of Rac1, rs10951982 and rs4720672, were identified as risk alleles associated with a higher risk of UC [43].
Two pathological molecular mechanisms of SRC were revealed in DSS-induced UC rats. SRC can be upregulated by Calcium/Ask1/MKK7/JNK2 signaling and induces Tyr-phosphorylation of occludin, ZO-1, E-cadherin and β-catenin, which disrupts tight junctions in the intestinal epithelium and leads to the epithelial barrier dysfunction [44]. Furthermore, SRC binds to and activates PIK3R1 (p85), a driver of PI3K signaling, resulting in activation of the NF-κB signaling cascade, which contributes to persistent inflammatory responses in injured intestinal tissues [45].
POLR2A is the largest catalytic subunit of RNA polymerase II (pol II) and is contained in Module 1. RNA pol II catalyzes various aspects of transcriptional events, including pre-mRNA splicing, which is a major step of mRNA processing [46, 47]. Enrichment analysis of Module 2 showed that DEGs in this module were mainly associated with RNA splicing and the spliceosome. Inflammation has been reported to perturb intra-cellular processes and lead to alternative pre-mRNA splicing in IECs, which alters the function, location or expression of a variety of proteins and promotes UC progression [48, 49].
A total of 24 drugs approved by FDA were predicted to have potential effects on four hub DEGs SRC, MAPK1, RAC1 and CDC42 and some of these drugs were confirmed to inhibit UC progression. MAPK1 is predicted to be targeted by Arsenic trioxide (As2O3) and ursodeoxycholic acid (ursodiol). In UC model rats induced by acetic acid or 2,4,6-trinitrobenzene sulfonic acid (TNBS), As2O3 treatment ameliorates colon inflammation through the reduction of inflammatory cells and oxidative stress [50, 51]. Ursodiol is found to protect epithelial barrier function by preventing the release of proinflammatory cytokines from IECs in DSS and TNBS-induced UC rats [52, 53]. Besides, the use of ursodiol is associated with a low risk of neoplasia in UC patients with primary sclerosing cholangitis [54, 55]. As an important tyrosine kinase, SRC is predicted to be targeted by several kind of tyrosine kinase inhibitors (TKIs). An SRC-specific TKI dasatinib shows significant anti-inflammatory effects in acetic acid-induced UC rats [56]. In addition to inhibiting the immune response, dasatinib can also protect the intestinal epithelial barrier of acetic acid-induced UC rats by decreasing the colonic vascular permeability [57]. Besides, the combination of dasatinib with TOP1210, a narrow spectrum kinase inhibitor that can inhibit SRC activity, exhibit improved potent inhibitory effects on pro-inflammatory cytokine release and immune response in UC [58].
The CIBERSORT algorithm revealed differences in the immune cell infiltration characteristics between pediatric and adult UC. Significant differences in the proportions of naive and memory B cells were revealed. In a recent study, CD23 was identified as a biomarker of mature naive B cells in pediatric UC, and the percentage of CD23+ B cells in pediatric UC patients was increased compared to that in non-IBD (inflammatory bowel disease) control children [59]. In contrast, the numbers of CD27+ memory B cells were decreased in UC compared to healthy controls, indicating that this population of cells participates in the pathogenesis of UC [60]. Compared with adult UC samples, we identified higher percentages of naive B cells and lower percentages of memory B cells in pediatric UC samples. Thus, it can be speculated that this profile of B cell types contributes to a more severe phenotype compared to adult UC.
Changes in the proportion of different subtypes of T cells are closely related to the development of UC [59, 61, 62]. Here, we found higher proportions of resting CD4 memory T cells and regulatory T cells (Tregs) in pediatric UC samples, while the proportions of follicular helper T cells and γδ T cells were lower. Tregs are essential for maintaining T cell homeostasis and immunological tolerance. In UC, the Treg cell frequency is decreased, which contributes to intestinal immunodysregulation and inflammation [63, 64]. Follicular helper T cells and γδ T cells were found to be activated in UC, leading to the positive regulation of inflammatory processes and dysimmunity typical of UC [61, 62, 64,65,66,67]. In addition, we found a higher percentage of dendritic cells (DCs) in adult UC, which is consistent with reports that alterations in DC phenotypes in UC drive aberrant T-cell responses [68, 69]. Thus, dysregulation of T cells in adult UC seems to be more severe than that in pediatric UC; this phenomenon requires further exploration.
Inflammatory processes in UC can be sustained by inappropriate activation or inactivation of monocytes and macrophages [70]. Compared with adult UC samples, we found higher proportions of monocytes and M0 macrophages in pediatric UC samples, while the proportions of M2 macrophages were lower. M2 macrophages are decreased in UC compared to healthy controls, and recovery of this population can antagonize M1 macrophage responses to inhibit inflammation [71,72,73,74,75]. Lower proportions of M2 macrophages in pediatric UC may lead to more severe inflammatory processes compared to those in adult UC. Furthermore, higher proportions of mast cells were detected in pediatric, representing another dysregulated immune cell population in UC that are activated and involved in disease development [76, 77].
In conclusion, a total of 1700 DEGs were identified in our bioinformatics analysis of colon samples from pediatric and adult UC patients based on gene dataset GSE87473. CDC42, POLR2A, RAC1, PIK3R1, MAPK1 and SRC were identified as the hub DEGs and may play important roles in the differences between pediatric and adult UC. Furthermore, pathway enrichment analysis of DEGs revealed that inter-cellular items that relate to cell junctions, cell adhesion, actin cytoskeleton and transmembrane receptor signaling pathways and intra-cellular items that relate to the splicing, metabolism and localization of RNA were involved in the differences between pediatric and adult UC. Finally, we investigated immune cell infiltration characteristics in pediatric and adult UC and identified the significant differences.
Inevitably, there were some innate limitations in the present study. The sample size was relatively small, and further experiments were lacked to verify the results due to the lack of relevant samples. All in all, these DEGs, pathways and immune cell infiltration characteristics identified in this study may provide insights into the molecular mechanisms of the differences between pediatric and adult UC, which require further confirmation in future studies.
Availability of data and materials
The dataset generated and/or analyzed during the current study are available from the Gene Expression Omnibus repository GSE87473.
References
Sauer CG, Kugathasan S. Pediatric inflammatory bowel disease: highlighting pediatric differences in IBD. Med Clin North Am. 2010;94:35-52.
Van Limbergen J, Russell RK, Drummond HE, Aldhous MC, Round NK, Nimmo ER et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology. 2008;135:1114-1122.
Turner D, Walsh CM, Benchimol EI, Mann EH, Thomas KE, Chow C et al. Severe paediatric ulcerative colitis: incidence, outcomes and optimal timing for second-line therapy. Gut. 2008;57:331-338.
Duricova D, Leroyer A, Savoye G, Sarter H, Pariente B, Aoucheta D et al. Extra-intestinal manifestations at diagnosis in paediatric- and elderly-onset ulcerative colitis are associated with a more severe disease outcome: a population-based study. J Crohns Colitis. 2017;11:1326-1334.
Malaty HM, Mehta S, Abraham B, Garnett EA, Ferry GD. The natural course of inflammatory bowel disease-indeterminate from childhood to adulthood: within a 25 year period. Clin Exp Gastroenterol. 2013;6:115-121.
Aloi M, D'Arcangelo G, Pofi F, Vassallo F, Rizzo V, Nuti F et al. Presenting features and disease course of pediatric ulcerative colitis. J Crohns Colitis. 2013;7:e509-515.
Dan T, Levine A, Escher JC, Griffiths AM, Ruemmele FM. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr. 2012;55:340-361.
Ruemmele FM, Turner D. Differences in the management of pediatric and adult onset ulcerative colitis--lessons from the joint ECCO and ESPGHAN consensus guidelines for the management of pediatric ulcerative colitis. J Crohns Colitis. 2014;8:1-4.
Ouahed J, Gordon W, Canavan JB, Zhou H, Du S, von Schack D et al. Mucosal gene expression in pediatric and adult patients with ulcerative colitis permits modeling of ideal biopsy collection strategy for transcriptomic analysis. Inflamm Bowel Dis. 2018;24:2565-2578.
Li K, Strauss R, Ouahed J, Chan D, Telesco SE, Shouval DS et al. Molecular comparison of adult and pediatric ulcerative colitis indicates broad similarity of molecular pathways in disease tissue. J Pediatr Gastroenterol Nutr. 2018;67:45-52.
Savic Mlakar A, Hojsak I, Jergovic M, Vojvoda Parcina V, Babic Z, Troskot B et al. Comparison of cytokine and efflux transporter expression in pediatric versus adult-onset ulcerative colitis. J Pediatr Gastroenterol Nutr. 2017;64:943-948.
Tatiya-Aphiradee N, Chatuphonprasert W, Jarukamjorn K. Immune response and inflammatory pathway of ulcerative colitis. J Basic Clin Physiol Pharmacol. 2018;30:1-10.
Neurath MF. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat Immunol. 2019;20:970-979.
Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3-10.
Liu Z, Meng J, Li X, Zhu F, Liu T, Wu G et al. Identification of hub genes and key pathways associated with two subtypes of diffuse large B-cell lymphoma based on gene expression profiling via integrated bioinformatics. Biomed Res Int. 2018;2018:3574534.
Li X, Liu Z, Mi M, Zhang C, Xiao Y, Liu X et al. Identification of hub genes and key pathways associated with angioimmunoblastic T-cell lymphoma using weighted gene co-expression network analysis. Cancer Manag Res. 2019;11:5209-5220.
Darbeheshti F, Rezaei N, Amoli MM, Mansoori Y, Tavakkoly Bazzaz J. Integrative analyses of triple negative dysregulated transcripts compared with non-triple negative tumors and their functional and molecular interactions. J Cell Physiol. 2019;234:22386-22399.
Zhang J, Zhou YJ, Yu ZH, Chen AX, Yu Y, Wang X et al. Identification of core genes and clinical roles in pregnancy-associated breast cancer based on integrated analysis of different microarray profile datasets. Biosci Rep. 2019;39.
Kalamohan K, Periasamy J, Bhaskar Rao D, Barnabas GD, Ponnaiyan S, Ganesan K. Transcriptional coexpression network reveals the involvement of varying stem cell features with different dysregulations in different gastric cancer subtypes. Mol Oncol. 2014;8:1306-1325.
Qin G, Mallik S, Mitra R, Li A, Jia P, Eischen CM et al. MicroRNA and transcription factor co-regulatory networks and subtype classification of seminoma and non-seminoma in testicular germ cell tumors. Sci Rep. 2020;10:852.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453-457.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41:D991-995.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607-d613.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-2504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2.
Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics (Oxford, England). 2008;24:282-284.
Cotto KC, Wagner AH, Feng YY, Kiwala S, Coffman AC, Spies G et al. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46:D1068-d1073.
Gao W, Wang C, Yu L, Sheng T, Wu Z, Wang X et al. Chlorogenic acid attenuates dextran sodium sulfate-induced ulcerative colitis in mice through MAPK/ERK/JNK pathway. Biomed Res Int. 2019;2019:6769789.
Lv Q, Qiao SM, Xia Y, Shi C, Xia YF, Chou GX et al. Norisoboldine ameliorates DSS-induced ulcerative colitis in mice through induction of regulatory T cells in colons. Int Immunopharmacol. 2015;29:787-797.
Cai L, Li X, Geng C, Lei X, Wang C. Molecular mechanisms of somatostatin-mediated intestinal epithelial barrier function restoration by upregulating claudin-4 in mice with DSS-induced colitis. Am J Physiol Cell Physiol. 2018;315:C527-C536.
Kim H, Banerjee N, Ivanov I, Pfent CM, Prudhomme KR, Bisson WH et al. Comparison of anti-inflammatory mechanisms of mango (Punica Granatum L.) and pomegranate (Punica Granatum L.) in a preclinical model of colitis. Mol Nutr Food Res. 2016;60:1912-1923.
Li X, Cai L, Xu H, Geng C, Lu J, Tao L et al. Somatostatin regulates NHE8 protein expression via the ERK1/2 MAPK pathway in DSS-induced colitis mice. Am J Physiol Gastrointest Liver Physiol. 2016;311:G954-G963.
Shi L, Lin Q, Yang T, Nie Y, Li X, Liu B et al. Oral administration of Lentinus edodes beta-glucans ameliorates DSS-induced ulcerative colitis in mice via MAPK-Elk-1 and MAPK-PPARgamma pathways. Food Funct. 2016;7:4614-4627.
You BH, Chae HS, Song J, Ko HW, Chin YW, Choi YH. alpha-Mangostin ameliorates dextran sulfate sodium-induced colitis through inhibition of NF-kappaB and MAPK pathways. Int Immunopharmacol. 2017;49:212-221.
Babbin BA, Jesaitis AJ, Ivanov AI, Kelly D, Laukoetter M, Nava P et al. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J Immunol. 2007;179:8112-8121.
Schlegel N, Meir M, Spindler V, Germer CT, Waschke J. Differential role of Rho GTPases in intestinal epithelial barrier regulation in vitro. J Cell Physiol. 2011;226:1196-1203.
Fu Y, Galan JE. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature. 1999;401:293-297.
Tang WJ, Peng KY, Tang ZF, Wang YH, Xue AJ, Huang Y. MicroRNA-15a—cell division cycle 42 signaling pathway in pathogenesis of pediatric inflammatory bowel disease. World J Gastroenterol. 2018;24:5234-5245.
Birkl D, Quiros M, Garcia-Hernandez V, Zhou DW, Brazil JC, Hilgarth R et al. TNFalpha promotes mucosal wound repair through enhanced platelet activating factor receptor signaling in the epithelium. Mucosal Immunol. 2019;12:909-918.
Kato T, Suzuki K, Okada S, Kamiyama H, Maeda T, Saito M et al. Aberrant methylation of PSD disturbs Rac1-mediated immune responses governing neutrophil chemotaxis and apoptosis in ulcerative colitis-associated carcinogenesis. Int J Oncol. 2012;40:942-950.
Wertheimer E, Kazanietz MG. Rac1 takes center stage in pancreatic cancer and ulcerative colitis: quantity matters. Gastroenterology. 2011;141:427-430.
Muise AM, Walters T, Xu W, Shen-Tu G, Guo CH, Fattouh R et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633-641.
Samak G, Chaudhry KK, Gangwar R, Narayanan D, Jaggar JH, Rao R. Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J. 2015;465:503-515.
Seok Yang W, Lee J, Woong Kim T, Hye Kim J, Lee S, Hee Rhee M et al. Src/NF-kappaB-targeted inhibition of LPS-induced macrophage activation and dextran sodium sulphate-induced colitis by Archidendron clypearia methanol extract. J Ethnopharmacol. 2012;142:287-293.
Saldi T, Cortazar MA, Sheridan RM, Bentley DL. Coupling of RNA polymerase ii transcription elongation with pre-mRNA splicing. J Mol Biol. 2016;428:2623-2635.
Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T et al. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014;28:2663-2676.
Hasler R, Kerick M, Mah N, Hultschig C, Richter G, Bretz F et al. Alterations of pre-mRNA splicing in human inflammatory bowel disease. Eur J Cell Biol. 2011;90:603-611.
Thoo L, Noti M, Krebs P. Keep calm: the intestinal barrier at the interface of peace and war. Cell Death Dis. 2019;10:849.
Moulahoum H, Boumaza BMA, Ferrat M, Bounaama A, Djerdjouri B. Arsenic trioxide ameliorates murine colon inflammation through inflammatory cell enzymatic modulation. Naunyn Schmiedeberg's Arch Pharmacol. 2019;392:259-270.
Singer M, Trugnan G, Chelbi-Alix MK. Arsenic trioxide reduces 2,4,6-trinitrobenzene sulfonic acid-induced murine colitis via nuclear factor-kappaB down-regulation and caspase-3 activation. Innate immunity. 2011;17:365-374.
Ward JBJ, Lajczak NK, Kelly OB, O'Dwyer AM, Giddam AK, Ni Gabhann J et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol. 2017;312:G550-g558.
Martinez-Moya P, Romero-Calvo I, Requena P, Hernandez-Chirlaque C, Aranda CJ, Gonzalez R et al. Dose-dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol. 2013;15:372-380.
Kim HS. Prevention of colon cancer with ursodiol in ulcerative colitis. Inflamm Bowel Dis. 2001;7:279-280.
Croog VJ, Ullman TA, Itzkowitz SH. Chemoprevention of colorectal cancer in ulcerative colitis. Int J Colorectal Dis. 2003;18:392-400.
Can G, Ayvaz S, Can H, Karaboga I, Demirtas S, Aksit H et al. The efficacy of tyrosine kinase inhibitor dasatinib on colonic mucosal damage in murine model of colitis. Clin Res Hepatol Gastroenterol. 2016;40:504-516.
Tolstanova HM, Khomenko TA, Ostapchenko LI, Szabo S, Sandor Z. [The role of Src-tyrosine kinases in increasing vascular permeability during experimental ulcerative colitis]. Ukrains'kyi biokhimichnyi zhurnal (1999 ). 2010;82:117-122.
Biancheri P, Foster MR, Fyfe MC, MacDonald TT, Sirohi S, Solanke Y et al. Effect of narrow spectrum versus selective kinase inhibitors on the intestinal proinflammatory immune response in ulcerative colitis. Inflamm Bowel Dis. 2016;22:1306-1315.
Rabe H, Malmquist M, Barkman C, Ostman S, Gjertsson I, Saalman R et al. Distinct patterns of naive, activated and memory T and B cells in blood of patients with ulcerative colitis or Crohn's disease. Clin Exp Immunol. 2019;197:111-129.
Wang X, Jiang Y, Zhu Y, Zhang M, Li M, Wang H et al. Circulating memory B cells and plasmablasts are associated with the levels of serum immunoglobulin in patients with ulcerative colitis. J Cell Mol Med. 2016;20:804-814.
Wang X, Zhu Y, Zhang M, Hou J, Wang H, Jiang Y et al. The shifted balance between circulating follicular regulatory T cells and follicular helper T cells in patients with ulcerative colitis. Clin Sci (Lond). 2017;131:2933-2945.
Xue G, Zhong Y, Hua L, Zhong M, Liu X, Chen X et al. Aberrant alteration of follicular T helper cells in ulcerative colitis patients and its correlations with interleukin-21 and B cell subsets. Medicine (Baltimore). 2019;98:e14757.
Mohammadnia-Afrouzi M, Zavaran Hosseini A, Khalili A, Abediankenari S, Hosseini V, Maleki I. Decrease of CD4(+) CD25(+) CD127(low) FoxP3(+) regulatory T cells with impaired suppressive function in untreated ulcerative colitis patients. Autoimmunity. 2015;48:556-561.
Sang LX, Chang B, Zhu JF, Yang FL, Li Y, Jiang XF et al. Sodium selenite ameliorates dextran sulfate sodium-induced chronic colitis in mice by decreasing Th1, Th17, and gammadeltaT and increasing CD4(+)CD25(+) regulatory T-cell responses. World J Gastroenterol. 2017;23:3850-3863.
Wang S, Fan T, Yao L, Ma R, Yang S, Yuan F. Circulating follicular regulatory T cells could inhibit Ig production in a CTLA-4-dependent manner but are dysregulated in ulcerative colitis. Mol Immunol. 2019;114:323-329.
Tsuchiya T, Fukuda S, Hamada H, Nakamura A, Kohama Y, Ishikawa H et al. Role of gamma delta T cells in the inflammatory response of experimental colitis mice. J Immunol. 2003;171:5507-5513.
McVay LD, Li B, Biancaniello R, Creighton MA, Bachwich D, Lichtenstein G et al. Changes in human mucosal gamma delta T cell repertoire and function associated with the disease process in inflammatory bowel disease. Mol Med. 1997;3:183-203.
Mann ER, Bernardo D, Ng SC, Rigby RJ, Al-Hassi HO, Landy J et al. Human gut dendritic cells drive aberrant gut-specific t-cell responses in ulcerative colitis, characterized by increased IL-4 production and loss of IL-22 and IFNγ. Inflamm Bowel Dis. 2014;20:2299-2307.
Al-Hassi HO, Mann ER, Sanchez B, English NR, Peake STC, Landy J et al. Altered human gut dendritic cell properties in ulcerative colitis are reversed by Lactobacillus plantarum extracellular encrypted peptide STp. Mol Nutr Food Res. 2014;58:1132-1143.
Gren ST, Grip O. Role of monocytes and intestinal macrophages in Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2016;22:1992-1998.
Zhu Y, Li X, Chen J, Chen T, Shi Z, Lei M et al. The pentacyclic triterpene Lupeol switches M1 macrophages to M2 and ameliorates experimental inflammatory bowel disease. Int Immunopharmacol. 2016;30:74-84.
Lin Y, Yang X, Yue W, Xu X, Li B, Zou L et al. Chemerin aggravates DSS-induced colitis by suppressing M2 macrophage polarization. Cell Mol Immunol. 2014;11:355-366.
Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci USA. 2012;109:9517-9522.
Lissner D, Schumann M, Batra A, Kredel L-I, Kühl AA, Erben U et al. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm Bowel Dis. 2015;21:1297-1305.
Zhu W, Yu J, Nie Y, Shi X, Liu Y, Li F et al. Disequilibrium of M1 and M2 macrophages correlates with the development of experimental inflammatory bowel diseases. Immunol Investig. 2014;43:638-652.
Boeckxstaens G. Mast cells and inflammatory bowel disease. Curr Opin Pharmacol. 2015;25:45-49.
Casado-Bedmar M, Heil SDS, Myrelid P, Söderholm JD, Keita ÅV. Upregulation of intestinal mucosal mast cells expressing VPAC1 in close proximity to vasoactive intestinal polypeptide in inflammatory bowel disease and murine colitis. Neurogastroenterol Motil. 2019;31:e13503-e13503.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81760328).
Author information
Authors and Affiliations
Contributions
MX and YL wrote the manuscript. GC, CH and BK coordinated and directed the project. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiu, Mx., Liu, Ym., Chen, Gy. et al. Identifying Hub Genes, Key Pathways and Immune Cell Infiltration Characteristics in Pediatric and Adult Ulcerative Colitis by Integrated Bioinformatic Analysis. Dig Dis Sci 66, 3002–3014 (2021). https://doi.org/10.1007/s10620-020-06611-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-020-06611-w