
Abstract
The prevalence of nonalcoholic fatty liver disease has been rapidly increasing worldwide. It has become a leading cause of liver transplantation. Accumulating evidence suggests a significant role for gut microbiota in its development and progression. Here we review the effect of gut microbiota on developing hepatic fatty infiltration and its progression. Current literature supports a possible role for gut microbiota in the development of liver steatosis, inflammation and fibrosis. We also review the literature on possible interventions for NAFLD that target the gut microbiota.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) ranges from simple steatosis (SS) to nonalcoholic steatohepatitis (NASH). Twenty to 30 percent of adults with NAFLD develop NASH. A subgroup of NASH patients develop severe morbidities such as cirrhosis, hepatocellular carcinoma and liver failure [1–3]. Prevalence of suspected NAFLD has rapidly increased over the past 20 years and currently affects about 11 % of adolescents [4] and 20 % of all ages [2]. This rapid increase among obese subjects, independent of body mass index (BMI), suggests that potentially modifiable risk factors other than obesity may have a role [4].
The number of bacterial cells present in the mammalian gut is a continuum that ranges from 101 to 103 bacteria per gram of contents in the stomach and duodenum, to 104–107 in the jejunum and ileum, culminating in 1011–1012 in the colon [5]. The human intestinal microbiome (IM) has 150-fold more genes compared to the host [6]. Therefore, the IM has been referred to as the missing organ. The IM is essential for several physiological functions including but not limited to vitamin biosynthesis, bile acid degradation and complex carbohydrate digestion. It is also important for intestinal mucosal barrier (IB) integrity.
The liver receives 70 % of its blood supply from the portal vein, the direct venous outflow of the intestine. This anatomical connection makes the liver the first line of defense against gut-derived products such as antigens and toxins [7]. This strong connection between the intestine and liver is termed the gut-liver axis and has been linked to liver pathogenesis since first described in 1998 [8].
Gut Microbiota and Obesity
Obesity is a critical risk factor for NAFLD. Here we review the interaction of the gut microbiota and obesity.
Recent animal studies suggest a role for microbiota in the pathogenesis of obesity. Bäckhed et al. showed that germ-free (GF) mice were leaner than conventionally raised mice despite consuming more energy. GF mice were resistant to developing obesity when fed a high-fat and a high-sugar diet. Colonizing GF mice with cecal microbiota obtained from conventionally raised mice led to a 60 % increase in body fat content and increased insulin resistance (IR) within 14 days despite reduced food intake [9]. Also, microbiota from ob/ob mice had an increased capacity to harvest energy from the diet [10]. Interestingly, this property was transferable; GF lean mice gavaged with an obese-mouse microbiota developed increased total body fat compared to mice gavaged with lean-mouse microbiota [10]. Moreover, GF mice colonized with microbiota from a lean human twin developed lower body mass and adiposity compared with those colonized from the obese co-twin, despite comparable energy intake [11]. Although these studies show a role for fecal microbiota in obesity, the transplant includes other substances such as short chain fatty acids (SCFAs), bile acids, and possibly undigested or partially digested food that in the short term could affect body mass and adiposity.
In humans, the IM differs between obese and lean subjects. Firmicutes and Bacteroidetes are the most abundant distal gut phyla, constituting over 90 % of known phylogenetic categories [12, 13]. The Bacteroidetes/Firmicutes ratio is disturbed in obesity and NASH. When subjects were controlled for intake of medications such gastric acid suppressors, antibiotics, probiotics or prebiotics, increased Bacteroidetes were found in obese adults, children and pregnant women [14]. Le Chatelier and Cotillard et al. also showed that obese and overweight adults are more likely to have a low microbial gene count (LGC) than non-obese adults. LGC subjects were found to have increased Bacteroides, gain more weight over time and experience increased IR, dyslipidemia and elevated inflammatory markers. Although the authors in these two studies accounted for antibiotics, diet and other food supplements were not considered [15, 16]. When obese women were subjected to Roux-en-Y gastric bypass (RYGB) and achieved weight loss, their IM changed. This change included an increase in IM diversity with 58 new genera detected after surgery in all patients. There was an increase in Bacteroides and Alistipes (phylum: Bacteroidetes), decrease in Lactobacillus, Dorea and Blautia (phylum: Firmicutes), and increase in Escherichia (phylum: Proteobacteria) [17]. The results of this study need to be interpreted carefully since another factor that can affect IM such as antibiotics, probiotics and medical diseases was not considered.
IM affects energy harvest. The human genome lacks a variety of enzymes including glycoside hydrolases and polysaccharide lyases required to digest complex carbohydrates. Fascinatingly, IM compensates for this lack by encoding for these very enzymes [18, 19]. Obese subjects have an increased Bacteroidetes/Firmicutes ratio [20–24]. Bacteroidetes encode for higher levels of complex carbohydrate-digesting enzymes compared to Firmicutes [18]. This suggests that the microbiome of obese individuals might have the capacity to harvest more energy.
SCFAs (mainly acetate, butyrate and propionate) are the end products of polysaccharide digestion. Although complex carbohydrates are the main source for SCFAs, they are also produced from amino acids by reductive deamination [25]. Animal studies showed that SCFA supplementation prevents total parenteral nutrition-associated mucosal atrophy and increases the intestinal absorption of glucose [26, 27]. The SCFA concentration is 20 % higher in obese and overweight adults than in lean adults on a western diet [22]. For people living in developed countries, SCFAs produced in the colon contribute approximately 5–10 % of the energy requirements. This estimate was based on a typical British diet, where 50–60 g of carbohydrates (15 g fiber and 35–50 g sugar and starch) are fermented per day [28], and could be even higher in areas of the world where more dietary fiber is consumed. Dietary fiber intake in Africa is estimated to be seven times higher than in the UK [29]. SCFAs serve as a fuel source for colonocytes, with butyrate being preferred [30, 31]. For example, in vitro, about 70 % of oxygen consumed by human colonocytes was the result of butyrate oxidation [32].
The production of SCFAs by colonic microbiota is relevant to obesity as they lead to increased calorie extraction and glucose absorption from the diet. On the other hand, SCFAs increase anorexigenic peptides. For example, in mice, SCFA supplementation was shown to protect against high-fat-diet (HFD)-induced obesity and IR through induction of gut hormones and subsequent reduction of food intake [33]. SCFAs have a similar effect on gut hormones in humans, leading to attenuation of appetite. Obese adults were blindly randomized to receive either inulin-propionate ester in sachets or inulin without propionate sachets (controls) at a dose of 10 g/day for 24 weeks. The content of the sachets was bound to a carrier molecule to deliver it specifically to the colon. The inulin-propionate group showed a significant reduction in weight gain, liver fat content, improved dyslipidemia and liver function tests (LFTs) [34]. In the same report, the investigators showed that inulin propionate significantly increased the release of postprandial plasma peptide YY (PYY) and glucagon-like peptide-1 (GLP-1) from colonocytes in vitro [34]. PYY and GLP-1 regulate appetite [35, 36].
In summary, there appears to be a link between gut microbiota and obesity, and this raises the possibility that alterations in the IM might have an effect on the development and sustainability of obesity.
Gut Microbiota and Steatosis
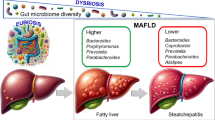
Hepatic steatosis is likely the result of many factors, including increased energy intake, de-novo lipogenesis and influx of free fatty acids, IR and dysbiosis. Dysbiosis is a deviation of the enteric microbiome from that found in healthy individuals, which has been shown in multiple studies (Table 1). However, there is no consistency or specificity to this difference. This is likely due to multiple factors that could influence the IM. Age, location where the study subjects reside as well as the method used to identify stool microbiota and diagnose NAFLD, biopsy or ultrasonography (US), could be partially responsible for this inconsistency. Also, not all the studies accounted for other confounders that affect the IM before stool collection such as antibiotics, acid-suppressing agents, probiotics, prebiotics, and whether there were attempts to control weight with specific dietary habits.
Factors affecting the IM composition such as HFD, choline-deficient diet (CDD), high-fructose diet, changes in bile acids and altered intestinal epithelium integrity have been linked to hepatic steatosis.
Mice fed with a saturated HFD based on palm oil exhibit increased hepatic steatosis, weight gain and reduced microbial diversity compared to mice fed an unsaturated fat-based diet of olive and safflower oil [37]. This observation suggests that the microbial changes associated with HFD are due to saturated fat. Yet, in mice fed HFD, the gut microbiota composition alters the lipid metabolism in the liver so steatosis can develop independently from obesity [38].
There is a relationship among choline, IM and NAFLD. Choline is derived from food such as red meat and eggs, but also can be biosynthesized [39]. Colonic bacteria can hydrolyze choline to form dimethylamine and trimethylamine, which are precursors of dimethylnitrosamine (DMN) [40–42]. DMN is a potent hepatotoxin, and carcinogen [43]. Choline is important in very low-density lipoprotein (VLDL) secretion by the liver, thus hepatic lipid homeostasis [39]. Both deficiency and over nutrition could have a negative impact on the liver. CDD is used to create a mouse model of NASH. Interestingly, an HFD reduces the bioavailability of choline, mimicking the effect of CDD when given to mice susceptible to impaired glucose homeostasis and NAFLD [44]. Therefore, alteration of the choline metabolism linked to steatosis could be due to HFD depleting choline. In humans, steatosis associated with parenteral nutrition is thought to be partly due to choline deficiency since supplementation with choline reverses the steatosis [45].
Phosphatidylethanolamine N-methyltransferase (PEMT) is important in endogenous phosphatidylcholine synthesis. PEMT polymorphism in adults and children influences susceptibility to choline deficiency-induced fatty liver [46]. Spencer et al. studied the effect of a choline-manipulated diet in 15 healthy females in relation to liver fat. They suggested that Gammaproteobacteria and Erysipelotrichi abundance, particularly when combined with the PEMT genotype, could predict choline deficiency-induced fatty liver [47].
High fructose consumption has been linked to NAFLD and its progression. In animal models, a high-fructose diet promotes NAFLD, alters microbiota composition, and increases endoplasmic reticulum stress and apoptotic activity [48, 49]. In humans, there are no such studies showing a link between high fructose consumption, microbiota and NAFLD. Two recent large clinical studies linked high fructose consumption with NAFLD histopathology, but the mechanism was not examined. The first study included 427 adults with NAFLD. Less steatosis and increased fibrosis were observed in those who had increased fructose intake [50]. Since fructose consumption was self-reported, serum uric acid, which is known to be a biologic marker for fructose [51, 52], was measured and found to be higher in subjects who reported increased fructose intake [50]. The second study included 149 children with NAFLD. Although histopathology did not correlate with self-reported fructose consumption, uric acid was significantly higher in NASH children compared to those with SS [53]. Thus, high fructose consumption might be associated with more pronounced NASH. However, more studies are needed to validate this association and examine whether there is any role for IM as shown in animal studies.
Bile acids might have a signaling function outside of the enterohepatic circulation, possibly involving glucose, lipid and energy homeostasis through activation of the farnesoid X receptor (FXR) and G-protein coupled receptor (TGR5). Animal studies showed that FXR is strongly expressed in the liver and intestine [54, 55]. FXR is known to have an important role in controlling hepatic de-novo lipogenesis, VLDL export and plasma triglyceride (TG) turnover [56]. TGR5 binds secondary bile acids and stimulates GLP-1 secretion, which plays a key role in glucose homeostasis [57]. Swann et al. [58] showed that the IM in rats affects the composition of the bile acid pool as well as the expression of genes controlled by FXR. They also observed that antibiotics could change the pool composition and lead to partial microbiota reduction [58]. HFD also affects bile acids. Mice fed with HFD have an altered bile acid composition that influences the gut microbial environment [59]. Human studies are lacking. However, FXR and TGR5 are activated by bile acids in humans [60–62], and they might serve similar functions. Collectively, these data show that bile acids can alter gut microbiota and vice versa, which could have a role in NAFLD.
IM homeostasis, including the abundance and diversity of phyla, families and genera, promotes a healthy IB, which is essential for liver health. Gut dysbiosis influences the integrity of the IB and exposes the liver to microbial products. For example, adults with biopsy-proven NAFLD (n = 35) compared to healthy controls (HC; n = 24) have small intestinal bacteria overgrowth (SIBO) and increased intestinal permeability with disturbed integrity of duodenal tight junctions. SIBO and intestinal permeability correlated with steatosis severity but not with NASH (n = 17) [63]. IB disruption in the pathogenesis of NAFLD is further supported by a recent study showing irregularly arranged duodenal microvilli and widened tight junctions in NAFLD adults compared to HC [64]. Similarly, intestinal permeability (measured by the lactulose/mannitol ratio) is increased in children with NAFLD compared to controls (children with asthma, upper respiratory tract infection and headache) [65]. Unlike the findings in adults, the only study performed on children showed that intestinal permeability was higher in children with NASH than in those exhibiting only steatosis. This association between increased permeability and NAFLD suggests a role for bacterial translocation exposing the liver to more microbial products and influencing the development and/or progression of NAFLD. The controversy with regards to gut permeability and its association with steatosis or NASH needs further study.
Thus, the microbiome is affected by many factors including diet and bile acids. It is possible that changes to the microbiota alter the intestinal barrier, allow intestinal contents access to the liver, and drive the development of steatosis and its progression to NASH.
Gut Microbiota and Inflammation
Inflammation in NASH is likely the result of many factors including, but not limited to, endogenous alcohol production, endotoxemia and inflammatory mediators. SCFAs might have antiinflammatory effects by helping to maintain a healthy IB.
Alcohol consumption is a known factor in alcoholic fatty liver disease and continued imbibing is associated with disease progression. Endogenously produced alcohol by IM may play a similar role in NASH. The enterobacteriaceae family including Escherichia exhibits mixed-acid fermentation, a major product of which is ethanol [66–68]. We reported that the abundance of Escherichia is significantly higher in adolescent NASH (n = 22) compared to obese subjects (n = 25) and HC (n = 16). Adolescents with NASH also had higher peripheral serum alcohol concentrations [23]. Higher ethanol metabolites are also observed in the stool of children with NAFLD diagnosed by US compared to obese with no ultrasound evidence of NAFLD or healthy lean children [69]. We showed that alcohol dehydrogenase (ADH) and aldehyde dehydrogenase are among the most highly upregulated genes in livers of adolescents with NASH [70]. A recent study reported that blood ethanol levels positively associate with IR in children with NAFLD [71]. In the same report hepatic ADH activity was shown to be significantly lower in ob/ob mice compared to controls. Based on the results in mice, the authors suggested that increased ethanol in NAFLD may result from insulin-dependent impairments of hepatic ADH activity [71]. This hypothesis goes against the observations that ADH activity is highly elevated in livers of adolescents with NASH [70], and the microbiome intervention reduces alcohol levels in ob/ob mice [72].
Similarly, a higher serum alcohol level is observed in adults with histology-proven NAFLD compared to HC [73]. One could expect an even higher alcohol level in the portal blood. Ethanol is shown to increase hepatocyte TG accumulation [74] and is a well-known source for generating reactive oxygen species (ROS), which can lead to steatohepatitis [75] and affect the IB integrity. Intestinal permeability measured by urinary excretion of the lactulose/mannitol ratio is higher in alcoholic steatohepatitis compared to subjects with no evidence of liver disease or HC [76]. The alcohol metabolite acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa [77]. Collectively, these data suggest that dysbiosis in NASH results in increased endogenous alcohol production, increased permeability of the IB and exposure of the liver to high concentrations of potentially harmful intestinal contents, similar to that seen in alcoholic steatohepatitis.
High energy intake is associated with increased levels of circulating LPS in humans and animals [78]. HFD might facilitate LPS uptake through elevated chylomicron production in intestinal epithelial cells [79]. Enhanced responsiveness to low-dose LPS, leading to liver injury and severe fibrosis, was observed in mice on an HFD compared to controls [80]. Taken together, these observations suggest a potential effect of HFD on altering the gut microbiota in favor of LPS-producing bacteria and enhancing the LPS impact on liver injury.
Animal studies suggest that endotoxemia alone might induce steatohepatitis [81–83]. In adults, some investigators have suggested a link between endotoxemia and NAFLD. There was a higher endotoxin level in all subjects with NAFLD compared to controls, and it was higher in NASH compared to subjects with steatosis [84–86]. However, one adult study showed that 17 % of NAFLD patients were free from endotoxemia [87]. We observed that endotoxemia is not necessarily found in NASH [88]. In our study of adolescents, none of the control subjects had a high endotoxin level. Only 28 % of obese individual and 42 % of NASH patients had high endotoxin levels; 71 % of obese and 58 % of NASH subjects had low endotoxin levels similar to HC [88]. This observation was supported by an earlier study showing that 22.5 % children with NAFLD had low levels of endotoxin [89]. Table 2 shows the human studies that looked at endotoxemia in NAFLD in further detail. To properly interpret these studies, it is necessary to consider the method used to measure LPS, how the patients were grouped, and whether there were any associated medical diseases that could affect the results. It is also important to consider that other confounders such as antibiotics and energy intake might have biased the results.
LPS and other microbial products are recognized by the Toll-like receptors (TLRs), which are expressed by liver cells, such as Kupffer cells (KCs), hepatic stellate cells (HSCs) and hepatocytes [90]. TLR-4 and TLR-9 receptors are the most studied and known receptors associated with liver injury [91, 92]. KCs produce inflammatory cytokines and enhance the activity of HSCs, which promote liver injury [91, 93, 94]. Cytokines, tumor necrosis factor α (TNF-α) and tumor growth factor-β1 (TGF-β1) have been strongly linked to more advanced NAFLD. TNF-α liver expression was shown to be increased in adults with NASH compared to obese adults with normal livers or only steatosis [95]. This observation was further supported by a larger scale study showing that hepatic TNF-α expression in adults with NAFLD (n = 92) is higher than in controls (n = 25) and even higher in NASH (n = 57) compared to subjects with only steatosis (n = 35) [80]. In children (n = 72), serum TNF-α correlated with the NAFLD activity score (NAS) [96]. TGF-β1 enhances HSC activation and promotes fibrogenesis [93, 94] by activating TLR-4, which is highly expressed by HSCs [91].
SCFAs might serve a protective role in inflammation. Butyrate was shown to have antiinflammatory effects on human monocytes by enhancing interleukin-10 (IL-10) secretion, inhibiting IL-12 and interferon-α (IFN-α) release [97]. Decreased fecal concentrations of butyrate and propionate have been reported in patients with ulcerative colitis (UC) [98]. UC is a chronic inflammatory bowel disease that affects the mucosal surface of the large intestine, mainly the sigmoid colon and rectum. SCFA administration in UC has beneficial clinical and laboratory outcomes [99–102]. These data suggest a role for SCFAs in maintaining IB integrity and a protective role against NAFLD by an antiinflammatory effect.
Gut Microbiota and Fibrosis
HSCs are the major producers of the fibrotic matrix [103–105]. These cells could be influenced by the IM through expression of TLR9 [92], which is the only known receptor for bacterial DNA [106, 107]. Hepatic fibrosis was shown to be significantly lower in TLR9−/− compared to TLR9+/+ in the bile duct ligation (BDL) mouse model [92, 108].
Dysbiosis can lead to the progression from normal liver to steatosis, inflammation and finally fibrosis. Fibrosis can occur independent of steatosis, and it is associated with dysbiosis itself. In mice, an HFD model subjected to BDL showed an increase in the percent of gram-negative bacteria, a reduced Bacteroidetes/Firmicutes ratio, complete disappearance of Bifidobacteriaceae (Phylum: Actinobacteria), a dramatic increase of gram-negative Proteobacteria, especially Enterobacteriaceae, and a reduction in microbial diversity in comparison to controls [109]. These microbiota changes were associated with decreased intrahepatic TG, increased hepatic mRNA expression of TLR4 and TLR9, increased HSCs activity and increased liver fibrosis. To assess the role of Proteobateria in fibrosis, De Minicus et al. transplanted selected gram-negative or -positive bacteria from the more fibrogenic HFD/BDL mouse model to the controls. The transplantation was done by oral gavage of donors’ cecal content. They found that control mice receiving the selected gram-negative flora showed a higher increase in liver injury compared to controls receiving the gram-positive fraction [109].
In humans, Boursier et al. recently showed that the type and function of gut microbiota in French adults with NAFLD (n = 57) is different based on the stage of fibrosis. Patients with stage 2 or greater had higher abundance of Bacteroides and Ruminococcus and a lower abundance of Prevotella compared to patients with fibrosis stage 0–1 [110].
Based on animal and human studies, microbiota dysbiosis could lead to fibrosis and contributes to its severity.
In summary, NASH patients have a different IM compared to healthy individuals. This altered microbiota can promote liver inflammation and fibrosis through different mechanisms, including but not limited to generating more alcohol and endotoxins that have increased access to the liver through disrupted IB. Overwhelming the liver with gut microbial products can activate an inflammatory cascade leading to hepatic injury. Manipulating this dysbiosis could have a preventive and therapeutic effect on NASH.
Interventions for NAFLD-Targeting Gut Microbiota
Diet
The diet can contribute to the contents of the gut microbiota. As discussed above, an HFD, high-energy diet, high-fructose diet, and decreased choline and its bioavailability lead to altered microbiota, which has been shown to be associated with NAFLD [37, 39, 48–50, 53, 78]. Moreover, an HFD and high-energy intake are linked to changes in the bile acid pool and increased LPS, which contribute to the development and promotion of NAFLD [59, 78]. Collectively, these data suggest that diet is the most potent tool to target NAFLD. Only 5 % loss of body weight can improve steatosis and 10 % can improve steatohepatitis [111]. Since it is not always possible to change dietary habits and maintain weight loss, other interventions that manipulate the IM might be beneficial.
Probiotics and Symbiotics
Table 3 shows the effect of probiotics and symbiotics on NAFLD. At this time, the data on the treatment of NAFLD with probiotics or symbiotics are not compelling. There is no standardized dose or strain or a clear understanding of treatment duration. There was no follow-up on any of the intervention groups. The trials focused only on the commercially available probiotics, and none of the trials targeted the specific microbiota depletion described in NAFLD. Most important, however, the content and concentrations of those preparations cannot be assured because they are exempt from FDA oversight (https://ods.od.nih.gov/About/DSHEA_Wording.aspx and http://thomas.loc.gov/cgi-bin/query/z?c103:S.784: See section 4).
Antibiotics
Antibiotics alter the IM by decreasing the growth of some bacteria and promoting the growth of others. There have been attempts to treat human metabolic syndrome and NAFLD with antibiotics. For example, 20 Dutch adults with metabolic syndrome were blindly randomized to either vancomycin (group A) or amoxicillin (group B) at a dose of 1500 mg/day for 7 days. Group A showed improved insulin sensitivity compared to no change in B. Group A showed a decrease of gram-positive bacteria (mainly Firmicutes) and a compensatory increase in gram-negative bacteria (mainly Proteobacteria) and decreased microbial diversity [112]. Regretfully, there was no assessment of the microbiota after discontinuation of the antibiotics. Decreased microbial diversity might not favor human health [15, 16]. Proteobacteria has been shown to be higher in NASH compared to HC [23]. Also increased Proteobacteria and reduced microbial diversity have been associated with increased liver fibrosis in mice [109].
A recent study examined the effect of oral rifaximin at a dose of 1200 mg/day for 28 days on Turkish adults with steatosis (n = 15) and NASH (n = 27). The NASH group showed a significant reduction in the mean BMI, alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyltransferase (GGT), low-density lipoprotein (LDL), LPS and IL-10 compared to the baseline. However, the steatosis group showed a significant reduction only in ALT. In both groups, there was no significant change in the levels of blood glucose, insulin, cholesterol, TG, C-reactive protein (CRP), homeostasis model of insulin resistance (HOMA IR), TNF-α, IL-1, IL-6 and IL-12 levels [113]. Although there were some favorable laboratory results with treatment, it is hard to accept them as a result of rifaximin. This was an open-label, observational study with no placebo control. The results could be biased by other factors such as diet and exercise, which were not considered. More importantly, IM, which was proposed as the mechanism behind this effect, was not examined.
Thus, antibiotics do not appear to offer a promising role in targeting microbial alteration promoting NAFLD.
Bile Acids
In animal models, bile acids were shown to have antimicrobial activity and an effect on the gut microbiota composition [114]. They also preserve the integrity of the IB [115]. BDL leads to bacterial overgrowth, mucosal injury and bacterial translocation [116, 117]. These observations inspired some investigators to test the effect of bile acid derivatives as a potential adjuvant treatment for NASH. In adults, a multicenter, randomized, placebo-controlled trial tested the effect of obeticholic acid (OCA), an FXR agonist, on NASH histopathology for 72 weeks. Less than 50 % of the OCA group showed histological improvement that was evident in patients with diabetes. Weight, ALT, AST and GTT improved during OCA treatment but returned to baseline after it was discontinued [118]. Although the NAS improved, OCA does not seem to be a promising treatment since dyslipidemia and pruritus were major side effects. Dyslipidemia is not a newly described result of OCA as it was reported in an earlier study that examined the effect of only 6 weeks of OCA treatment of diabetic patients with presumed NAFLD [119].
Conclusion
Patients with NAFLD have specific enteric microbial alterations that could promote liver injury through multiple routes. Diet seems to be the driving force behind the development of such dysbiosis. There is an increase in the Enterobacteriaceae family (phylum: Proteobacteria), especially Escherichia, which produces alcohol and expresses endotoxins. Enterobacteriaceae is also shown to be the main bacteria associated with fibrosis in an HFD/BDL animal model. Proteobacteria is abundant in obese and NASH adolescents compared to healthy controls. It is also observed to be the only abundant phylum exhibiting a significant difference between the obese and NASH groups. There is an increase in the Bacteroidetes/Firmicutes ratio. There is depletion of certain taxa such as Bifidobacteriaceae, Lachnospiraceae, Veillonellaceae and Ruminococcaceae [23]. Although probiotics appear to be promising aids in the management of NAFLD, more effort is needed to target the specific NAFLD microbiota alteration, standardize the dose and determine the length of the intervention. Diet appears to be the overwhelming factor influencing the microbiome, and dietary change to reduce fat is the safest and most effective treatment for NASH.
Key Messages
-
Diet plays an essential role in NAFLD and contributes to its progression, possibly by its effect on the enteric microbial composition.
-
Stool microbiota in obese and NAFLD patients is different from that in healthy lean individuals.
-
Intestinal microbiota composition affects the intestinal barrier function, which is important for liver health.
-
Diet is the strongest tool to overcome NAFLD.
-
Targeting NAFLD-specific microbial alterations is a promising adjunct intervention.
References
Adams LA, Lymp JF, St Sauver J, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121.
Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285.
Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643–654. (e641–e649; quiz e639–e640).
Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J Pediatr. 2013;162:e491.
Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65.
Compare D, Coccoli P, Rocco A, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22:471–476.
Pereira SP, Rhodes JM, Campbell BJ, et al. Biliary lactoferrin concentrations are increased in active inflammatory bowel disease: a factor in the pathogenesis of primary sclerosing cholangitis? Clin Sci. 1998;95:637–644.
Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031.
Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214.
Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638.
Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Zhu L, Baker RD, Baker SS. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr Res. 2015;77:245–251.
Le Chatelier E, Nielsen T, Qin J, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546.
Cotillard A, Kennedy SP, Kong LC, et al. Dietary intervention impact on gut microbial gene richness. Nature. 2013;500:585–588.
Kong LC, Tap J, Aron-Wisnewsky J, et al. Gut microbiota after gastric bypass in human obesity: increased richness and associations of bacterial genera with adipose tissue genes. Am J Clin Nutr. 2013;98:16–24.
El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11:497–504.
Xu J, Bjursell MK, Himrod J, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299:2074–2076.
Kakiyama G, Pandak WM, Gillevet PM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58:949–955.
Collado MC, Isolauri E, Laitinen K, Salminen S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr. 2008;88:894–899.
Schwiertz A, Taras D, Schafer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18:190–195.
Zhu L, Baker SS, Gill C, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609.
Wong VW, Tse CH, Lam TT, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—a longitudinal study. PLoS One. 2013;8:e62885.
Blachier F, Mariotti F, Huneau JF, Tome D. Effects of amino acid-derived luminal metabolites on the colonic epithelium and physiopathological consequences. Amino Acids. 2007;33:547–562.
Koruda MJ, Rolandelli RH, Bliss DZ, Hastings J, Rombeau JL, Settle RG. Parenteral nutrition supplemented with short-chain fatty acids: effect on the small-bowel mucosa in normal rats. Am J Clin Nutr. 1990;51:685–689.
Tappenden KA, Thomson AB, Wild GE, McBurney MI. Short-chain fatty acid-supplemented total parenteral nutrition enhances functional adaptation to intestinal resection in rats. Gastroenterology. 1997;112:792–802.
McNeil NI. The contribution of the large intestine to energy supplies in man. Am J Clin Nutr. 1984;39:338–342.
Bingham S, Cummings JH, McNeil NI. Intakes and sources of dietary fiber in the British population. Am J Clin Nutr. 1979;32:1313–1319.
Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81:1031–1064.
Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40:235–243.
Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980;21:793–798.
Lin HV, Frassetto A, Kowalik EJ Jr, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS One. 2012;7:e35240.
Chambers ES, Viardot A, Psichas A, et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. 2015;64:1744–1754.
Batterham RL, Cowley MA, Small CJ, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–654.
Batterham RL, Cohen MA, Ellis SM, et al. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med. 2003;349:941–948.
de Wit N, Derrien M, Bosch-Vermeulen H, et al. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Gastrointest Liver Physiol. 2012;303:G589–G599.
Le Roy T, Llopis M, Lepage P, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. 2013;62:1787–1794.
Vance DE. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr Opin Lipidol. 2008;19:229–234.
Zeisel SH, DaCosta KA, Fox JG. Endogenous formation of dimethylamine. Biochem J. 1985;232:403–408.
Zeisel SH, daCosta KA, LaMont JT. Mono-, di- and trimethylamine in human gastric fluid: potential substrates for nitrosodimethylamine formation. Carcinogenesis. 1988;9:179–181.
Zeisel SH, daCosta KA, Youssef M, Hensey S. Conversion of dietary choline to trimethylamine and dimethylamine in rats: dose-response relationship. J Nutr. 1989;119:800–804.
Haggerty HG, Holsapple MP. Role of metabolism in dimethylnitrosamine-induced immunosuppression: a review. Toxicology. 1990;63:1–23.
Dumas ME, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. 2006;103:12511–12516.
Buchman AL, Dubin MD, Moukarzel AA, et al. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology. 1995;22:1399–1403.
Song J, da Costa KA, Fischer LM, et al. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD). FASEB J. 2005;19:1266–1271.
Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140:976–986.
Collison KS, Saleh SM, Bakheet RH, et al. Diabetes of the liver: the link between nonalcoholic fatty liver disease and HFCS-55. Obesity. 2009;17:2003–2013.
Tappy L, Le KA. Does fructose consumption contribute to non-alcoholic fatty liver disease? Clin Res Hepatol Gastroenterol. 2012;36:554–560.
Abdelmalek MF, Suzuki A, Guy C, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1961–1971.
Cox CL, Stanhope KL, Schwarz JM, et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr Metab (Lond). 2012;9:68.
Lin WT, Huang HL, Huang MC, et al. Effects on uric acid, body mass index and blood pressure in adolescents of consuming beverages sweetened with high-fructose corn syrup. Int J Obes (Lond). 2013;37:532–539.
Vos MB, Colvin R, Belt P, et al. Correlation of vitamin E, uric acid, and diet composition with histologic features of pediatric NAFLD. J Pediatr Gastroenterol Nutr. 2012;54:90–96.
Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799.
Li Z, Kruijt JK, van der Sluis RJ, Van Berkel TJ, Hoekstra M. Nuclear receptor atlas of female mouse liver parenchymal, endothelial, and Kupffer cells. Physiol Genomics. 2013;45:268–275.
Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M. Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis. 2010;28:220–224.
Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177.
Swann JR, Want EJ, Geier FM, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA. 2011;108:4523–4530.
Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–108.
Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440.
Fuchs M. Non-alcoholic Fatty liver disease: the bile Acid-activated farnesoid x receptor as an emerging treatment target. J Lipids. 2012;2012:934396.
Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67.
Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887.
Jiang W, Wu N, Wang X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep. 2015;5:8096.
Giorgio V, Miele L, Principessa L, et al. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig Liver Dis. 2014;46:556–560.
Dawes EA, Foster SM. The formation of ethanol in Escherichia coli. Biochim Biophys Acta. 1956;22:253–265.
Paege LM, Gibbs M. Anaerobic dissimilation of glucose-C14 by Escherichia coli. J Bacteriol. 1961;81:107–110.
Brooks JB, Basta MT, el Kholy AM. Studies of metabolites in diarrheal stool specimens containing Shigella species by frequency-pulsed electron capture gas-liquid chromatography. J Clin Microbiol. 1985;21:599–606.
Michail S, Lin M, Frey MR, et al. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol Ecol. 2015;91:1–9.
Baker SS, Baker RD, Liu W, Nowak NJ, Zhu L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS One. 2010;5:e9570.
Engstler AJ, Aumiller T, Degen C, et al. Insulin resistance alters hepatic ethanol metabolism: studies in mice and children with non-alcoholic fatty liver disease. Gut. 2015. doi:10.1136/gutjnl-2014-308379.
Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology. 2000;119:1340–1347.
Volynets V, Kuper MA, Strahl S, et al. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig Dis Sci. 2012;57:1932–1941.
Hartmann P, Chen WC, Schnabl B. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front Physiol. 2012;3:402.
Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19.
Keshavarzian A, Holmes EW, Patel M, Iber F, Fields JZ, Pethkar S. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207.
Basuroy S, Sheth P, Mansbach CM, Rao RK. Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and l-glutamine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G367–G375.
Amar J, Burcelin R, Ruidavets JB, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87:1219–1223.
Laugerette F, Vors C, Geloen A, et al. Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J Nutr Biochem. 2011;22:53–59.
Imajo K, Fujita K, Yoneda M, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16:44–54.
Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772.
Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185.
Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–1059.
Ruiz AG, Casafont F, Crespo J, et al. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg. 2007;17:1374–1380.
Harte AL, da Silva NF, Creely SJ, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm. 2010;7:15.
Verdam FJ, Rensen SS, Driessen A, Greve JW, Buurman WA. Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J Clin Gastroenterol. 2011;45:149–152.
Thuy S, Ladurner R, Volynets V, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452–1455.
Yuan J, Baker SS, Liu W, et al. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 2014;29:1292–1298.
Alisi A, Manco M, Devito R, Piemonte F, Nobili V. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. 2010;50:645–649.
Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720.
Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332.
Gabele E, Muhlbauer M, Dorn C, et al. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun.. 2008;376:271–276.
Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84:1780–1785.
Rivera CA, Bradford BU, Hunt KJ, et al. Attenuation of CCl(4)-induced hepatic fibrosis by GdCl(3) treatment or dietary glycine. Am J Physiol Gastrointest Liver Physiol. 2001;281:G200–G207.
Crespo J, Cayon A, Fernandez-Gil P, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158–1163.
Manco M, Marcellini M, Giannone G, Nobili V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;127:954–960.
Saemann MD, Bohmig GA, Osterreicher CH, et al. Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 2000;14:2380–2382.
Takaishi H, Matsuki T, Nakazawa A, et al. Imbalance in intestinal microflora constitution could be involved in the pathogenesis of inflammatory bowel disease. Int J Med Microbiol. 2008;298:463–472.
Scheppach W. Treatment of distal ulcerative colitis with short-chain fatty acid enemas. A placebo-controlled trial. German-Austrian SCFA Study Group. Dig Dis Sci. 1996;41:2254–2259.
Luhrs H, Gerke T, Muller JG, et al. Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol. 2002;37:458–466.
Vernia P, Annese V, Bresci G, et al. Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur J Clin Investig. 2003;33:244–248.
Hamer HM, Jonkers DM, Vanhoutvin SA, et al. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin Nutr. 2010;29:738–744.
Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400.
Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–1473.
Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680.
Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95.
Harry D, Anand R, Holt S, et al. Increased sensitivity to endotoxemia in the bile duct-ligated cirrhotic rat. Hepatology. 1999;30:1198–1205.
De Minicis S, Rychlicki C, Agostinelli L, et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738–1749.
Boursier J, Mueller O, Barret M, et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2015. doi:10.1002/hep.28356.
Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609.
Vrieze A, Out C, Fuentes S, et al. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol. 2014;60:824–831.
Gangarapu V, Ince AT, Baysal B, et al. Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2015;27:840–845.
Islam KB, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141:1773–1781.
Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472.
Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–3925.
Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283–1292.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965.
Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–582. (e571).
Mouzaki M, Comelli EM, Arendt BM, et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–127.
Raman M, Ahmed I, Gillevet PM, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2013;11:e861–e863.
Malaguarnera M, Vacante M, Antic T, et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57:545–553.
Aller R, De Luis DA, Izaola O, et al. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15:1090–1095.
Wong VW, Won GL, Chim AM, et al. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol. 2013;12:256–262.
Eslamparast T, Poustchi H, Zamani F, Sharafkhah M, Malekzadeh R, Hekmatdoost A. Synbiotic supplementation in nonalcoholic fatty liver disease: a randomized, double-blind, placebo-controlled pilot study. Am J Clin Nutr. 2014;99:535–542.
Vajro P, Mandato C, Licenziati MR, et al. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J Pediatr Gastroenterol Nutr. 2011;52:740–743.
Alisi A, Bedogni G, Baviera G, et al. Randomised clinical trial: the beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2014;39:1276–1285.
Acknowledgments
Work done in the authors’ laboratory is mainly supported by Grants from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (U01 DK061728 to S.S.B.), the Peter and Tommy Fund, Inc., Buffalo, NY (to S.S.B. and L.Z.) and a departmental start-up fund (to L.Z.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors had any relevant personal or financial conflicts of interest.
Rights and permissions
About this article

Cite this article
Abdou, R.M., Zhu, L., Baker, R.D. et al. Gut Microbiota of Nonalcoholic Fatty Liver Disease. Dig Dis Sci 61, 1268–1281 (2016). https://doi.org/10.1007/s10620-016-4045-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-016-4045-1