
Abstract
Background
MicroRNAs (miRNAs) are a class of small (19–25 nucleotides) noncoding RNAs that regulate the expressions of a wide variety of genes, including some involved in cancer development. Some recent studies show that DNA methylation contributes to down-regulation of microRNA-375 (miR-375) during tumorigenesis. Whether or not down-regulation of miR-375 also exists in esophageal cancer is unknown.
Aim
Our aim was to test the hypothesis that down-regulation of miR-375 also exists in esophageal cancer.
Methods
Expression of levels of miR-375 were examined using real-time PCR on paired esophageal cancer and adjacent non-cancerous tissues. The methylation status is detected by methylation specific-PCR (MSP).
Results
The results show that miR-375 is downregulated by hypermethylation of the promoter in esophageal cancer tissues. Epigenetic silencing of miR-375 induced an up-regulation of its targets, 3-phosphoinositide-dependent protein kinase-1 (PDK1). Restoration of the miR-375 expression in esophageal cancer cell lines downregulated the PDK1 expression. Furthermore, the miR-375 expression was found to be inversely correlated with PDK1 expression in esophageal cancer.
Conclusion
Thus, miR-375 is frequently down-regulated in esophageal cancer and is a negative regulator of PDK1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs are 19–25-nucleotides regulatory non-protein coding RNA molecules that usually function as endogenous repressors of target genes, resulting in mRNA degradation or inhibition of translation [1, 2]. The resulting gene repression occurs by multiple mechanisms including enhanced mRNA degradation and translational repression [3]. Developmental timing, cell death, proliferation, hematopoiesis, insulin secretion, and the immune response are just a few examples of critical biological events that depend on faithful miRNA expression [4]. Studies have shown that profiles of miRNA expression differ between normal and tumour tissues, which vary among tumour types [4, 5]. The downregulation of miRNA subsets implies a tumor-suppressor function, which is often observed in tumour development [6, 7]. For example, overexpression of miR-375 significantly inhibited gastric cancer cell proliferation in vitro and in vivo. Forced expression of miR-375 in gastric cancer cells significantly reduced the protein level of Janus kinase 2 (JAK2) and repressed the activity of a luciferase reporter carrying the 3′-untranslated region of JAK2, which was abolished by mutation of the predicted miR-375-binding site, indicating that JAK2 may be a miR-375 target gene [8].
Studies have extensively shown that hypermethylation of gene promoter is a frequent mechanism of silencing and associated with the prognosis of the diseases and response to therapy in patients with cancer. In the present study, we found that miR-375 is downregulated by hypermethylation of the promoter in esophageal cancer tissues. Epigenetic silencing of miR-375 induced an up-regulation of its targets, 3-phosphoinositide-dependent protein kinase-1 (PDK1). Restoration of the miR-375 expression in esophageal cancer cell lines downregulated the PDK1 expression. Furthermore, we found that the inhibition of anchorage-independent growth and induction of apoptosis after re-expression of miR-375 was partially through suppression of the PDK1/Akt pathway.
Materials and Methods
Patients and Cell Line
Human esophageal cancer samples were collected from surgical specimens from 40 patients with esophageal cancer at the Department of Gastroenterology, the First Affiliated Hospital, Zhengzhou University, Zhengzhou, China. Non-tumor samples from the macroscopic tumor margin were isolated at the same time and used as the matched adjacent non-neoplastic tissues. All the samples were divided into two parts. Tissue samples were collected, immediately snap frozen in liquid nitrogen, and stored at −80°C until RNA extraction. Informed consent was obtained from all patients. All patients obtained a confirmed diagnosis of esophageal cancer after resection. Five esophageal cancer cell lines (EC9706, EC109, HCT-15, TE-1 and EC-1) were all preserved in our laboratory and maintained in DMEM with 10% FBS.
Immunohistochemistry
Immunohistochemistry was performed by a two-step method using primary antibody including heat-induced antigen-retrieval procedures. Sections were incubated overnight at 37°C with primary antibody; after the primary antibody was washed off, the components of the Envision detection system were applied with an anti-mouse polymer (EnVision1/HRP/Mo, Dako, Glostrup, Denmark). The primary antibodies used were all mouse-antihuman monoclonal antibodies against PDK1 (1:100 dilution; Cell Signaling Technology). Negative controls were treated identically but with the primary antibody omitted. Immunoreactivity was evaluated independently by three researchers who were blinded to patient outcome. The percentage of positive tumor cells was determined by each observer, and the average of three scores was calculated. We randomly selected ten high-power fields; and counted 1,000 cells in each core. When the mean of percentage of positive cells is close to 0% or 100%, the standard deviation (SD) is close to 0, and when the mean is approximately 50% the SD is approximately 5%. Thus, the SD does not increase with the mean. The following categories were used for scoring: intensity of staining, none (0), mild (1), strong (2); percentage of the positive staining, <5% (0), 5–25% (1), 25–50% (2), >50% (3). Combining intensity and percentage staining resulted in the following score: 0–1, negative (−); 2–6, positive (+).
Methylation Specific-PCR
Genomic DNA (2 μg) was modified with sodium bisulfite using a EpiTect Bisulfite kit (Qiagen). Methylation status was analyzed by methylation specific-PCR (MSP). For the methylated allele, the sense primer was 5′-TTTTTTATTTTTCGCGTCGC G-3′ and the antisense primer was 5′-CCTAATTCCTAACCCTCCGCG-3′. For the unmethylated allele, the sense primer was 5′-GTTAGTGTTGGTTGTTGGATTG-3′ and the antisense primer was 5′-CCTCCAAACAAACACAACACA-3′. Hot-start PCR with a total cycle number of 30 was used in all MSP DNA amplifications. Denaturation and extension cycles were maintained for 30 and 45 s, respectively.
Real-Time qPCR
Total RNA was extracted from esophageal cancer or normal tissues using TRIZOL (Invitrogen) according to the manufacturer’s instructions. For miRNA qPCR, reverse transcription was performed using the QuantMir RT KIT (System Bioscience). In brief, 1 μg of RNA containing miRNA was polyadenylated by poly (A) polymerase and then reverse transcribed to cDNA using oligo-dT primers. The cDNA then serves as the template for SYBR real-time PCR using SYBR-green PCR Master Mix (Applied Biosystems). The miR-375-specific forward primer sequence was designed on the basis of miRNA sequences obtained from the miRBase database. Human U6 snRNA was used for normalization. For PDK1 mRNA qPCR, reverse transcription was carried out using Moloney murine leukemia virus reverse transcriptase (M-MLV RT, Promega). Following the protocol of the manufacturer, the amount of PDK1 expression, normalized to a human actin endogenous reference, is given by: 2−ΔΔCt. Real-time RT–PCR was repeated at least three times for each specimen and the mean was obtained.
Cell Transfection
The pre-miR-375 precursor molecule (miR-375 mimics), anti-miR-375 and negative control RNA-oligonucleotides were gained from Ambion corporation (Ambion, Austin, USA). The day before transfection, esophageal cancer cells were seeded in antibiotic free medium. Transfection of miRNAs was carried out using Lipofectamine 2000 in accordance with the manufacturer’s procedure (Invitrogen, California, USA). The level of miR-375 mimics expression in the esophageal cancer cells was assayed by real-time PCR.
Cell Growth Assay
Esophageal cancer cells were seeded in 96-well plates 1 day before transfection. After transfecting with miR-375, control oligo, the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) was used to determine relative cell growth.
Anchorage-Independent Growth
For anchorage-independent growth assay, 1 × 104 cells were plated in 0.3% low melting point agar/growth medium onto 6-cm dishes with a 0.6% agar underlay. After 4 weeks, colonies that were >1 mm in diameter were counted.
Evaluation of Apoptosis
Apoptosis was detected by flow cytometric analysis of Annexin V staining. Annexin V–FITC versus PI assay was performed as previously reported. Briefly, adherent cells were harvested and suspended in the Annexin-binding buffer (1 × 106 cells/ml). Thereafter, cells were incubated with Annexin V–FITC and PI for 15 min at room temperature in the dark and immediately analyzed by flow-cytometry. The data are presented as bi-parametric dot plots showing Annexin V–FITC green fluorescence versus PI red fluorescence.
Luciferase Activity Assay
A fragment of the wild-type (WT) PDK1 3′UTR containing the predicted miR-375 binding site was amplified by RT–PCR. Site-directed mutagenesis of the miR-375 target site was carried out using the Stratagene Quik-Change site-directed mutagenesis kit (Stratagene, Heidelberg, Germany). The construct was sequenced and named PDK-UTR-Mut. The pMIR-report luciferase vector was used for the construction of PDK-UTR or PDK-UTR-Mut plasmids (Ambion, USA). EC9706 and EC-1 cells were cultured in 24-well plates. In each well, 10 ng of phRL-TK renilla luciferase vector (Promega, USA) was co-transfected to normalize transfection efficiency. A sample of 500 ng of PDK-UTR or PDK-UTR-Mut plasmids together with 10 nM miR-375 mimics or negative control was also co-transfected. Transfection was done using Lipofectamine 2000 and Opti-MEM I reduced serum medium (Life Technologies, California, USA). Firefly luciferase activity was measured using the Dual luciferase assay kit (Promega). Normalized relative luciferase activity (RLA) was calculated as the following formula: RLA = [firefly luciferase]/[renilla luciferase].
Western Blot Analysis
Protein of treated cell lines was extracted by mammalian protein extraction reagent (Pierce, USA) supplemented with protease inhibitors cocktail (Sigma, USA). Then 50-μg protein samples were resolved by 10% SDS–PAGE and transferred to PVDF membranes. Autoradiograms were quantified by densitometry (Quantity One software; Bio-Rad, Hercules, CA). Actin-specific antibody was used for loading control. Mouse monoclonal anti-PDK1 (1:1,000, Cell Signaling Technology), anti-phosphorylated Akt antibody (Ser473; 1:1,000, Cell Signaling Technology) and mouse monoclonal anti-Actin (1:1,000, abcam, USA) were used.
Statistical Analysis
Expression of miR-375 in paired esophageal cancer and adjacent non-cancerous tissues were compared by paired t-test. The difference between the two groups in cell proliferation assay and luciferase reporter assay was analyzed by two-sided Student’s t-test. Data are expressed as the mean ± SD from at least three independent experiments. All P values are two-sided and a value of <0.05 was considered to be statistically significant.
Results
Involvement of DNA Methylation in Downregulation of miR-375
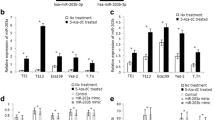
It was reported that the expression of miR-375 was regulated by CpG island methylation in breast cancer [9]; we determined whether this epigenetic mechanism also exists in esophageal cancer. The methylation of promoter was detected by methylation-specific PCR (MSP). We found that the miR-375 promoter was methylated in 30 of 40 (75%) cancer tissues and 2 of 40 (5%) in adjacent non-cancerous tissues (P < 0.01). Representative examples of the gel analysis of MSP are shown in Fig. 1a. Correlation of the promoter methylation data with miR-375 expression revealed a significantly lower mean expression level in the 30 tumors with promoter methylation as compared with the tumors without promoter methylation (Fig. 1b). The expression of miR-375 was further determined in a panel of five human esophageal cancer cell lines (EC9706, EC109, HCT-15, TE-1 and EC-1). The promoter of miR-375 was markedly methylated in all of the five esophageal cancer cell lines. To test that this promoter methylation was responsible for the downregulation of miR-375, esophageal cancer cell lines were treated with DAC (an inhibitor of the methylase enzyme, which can re-activate mRNA expression suppressed by methylation) for 48 h and performed RT–PCR to detect miR-375 expression. After treatment with DAC, we found the levels of miR-375 were induced in these esophageal cancer cell lines, suggesting that the miR-375 gene silencing is accounted for by hypermethylation. The expression levels of miR-375 were quantified by real-time PCR (Fig. 2a). The demethylation of miR-375 by DAC in these esophageal cancer cells was verified by use of MSP (Fig. 2b).

a Representative MSP results of miR-375 hypermethylation in esophageal cancer tissues. Case numbers are shown on top. M methylated primers, U unmethylated primers. b Relative miR-375 expression in 40 esophageal cancer tissues stratified by miR-375 promoter methylation. miR-375 levels and promoter methylation were determined by qRT-PCR and MSP, respectively. Depending on each tumor’s promoter methylation status, the cases were subdivided into two groups, i.e., unmethylated (n = 10 tumors) and methylated (n = 30 tumors). Note significantly lower mean miR-375 expression in the tumors with promoter methylation (P < 0.01, two-sided t test)

a Induction of miR-375 by treatment of esophageal cancer cell lines with DAC; levels of miR-375 were quantified by real-time PCR and normalized to U6. Graph represents means of three independent experiments ± standard deviations. Asterisk indicates the changes of miR-375 in these esophageal cancer cells after the treatment of DAC (P < 0.01). b The methylation status of miR-375 in esophageal cancer cell lines treated with or without DAC as determined by MSP. M methylated primers, U unmethylated primers
Ectopic miR-375 Expression Alters Esophageal Cancer Cell Growth, Apoptosis, and Anchorage-Independent Growth
Frequent downregulation of miR-375 in esophageal cancer cell lines and primary esophageal cancer tissues implies that miR-375 may have a role in esophageal cancer carcinogenesis. To prove this, the effect of ectopic expression of miR-375 on cell growth was investigated in two esophageal cancer cell lines (EC9706 and EC-1). We transfected 50 nM of miR-375 RNA oligonucleotides (Ambion) into EC9706 and EC-1 cells followed by analysis using the Kit-8 assay 48 h later. Scrambled oligonucleotides purchased from Ambion were used as a negative control. All transfections and Kit-8 assays were performed in triplicate in 96-well plates. The increased expression of miR-375 by ectopic miR-375 expression significantly inhibited the growth of EC9706 and EC-1 cells (data not shown). To further understand the mechanisms by which cell proliferation is affected, flow cytometry was performed to analyze the cell apoptosis percentage. We did transient transfections of miR-375 or negative control RNA-oligonucleotides transfections at a final concentration of 50 nM in the cancer cell lines. The exogenous expression of miR-375 in cells was confirmed by real-time PCR 48 h after transfection as described above (data not shown). We found that cells treated with miR-375 undergo a higher level of apoptosis than cells treated with negative control RNA-oligonucleotides (P < 0.001) (Fig. 3a). Next, we chose EC9706 cells for anchorage-independent growth assay, namely, soft agar assay, to detect the effect of miR-375 on colony formation ability. The results showed that the exogenous expression of miR-375 dramatically inhibited the anchorage-independent ability when compared to control (P < 0.001) (Fig. 3b).

a The apoptotic percentage of the cells with miR-375 precursor molecule or Mock was detected by flow cytometric analysis. The values above each bar represent the fraction of annexin V+/PI− and annexin V+/PI+. Values are expressed as the mean ± SD of three replicate experiments. *P < 0.001 compared with Mock. b Examples of colony assay after 4-week selection. Left bottom detailed images from the top panels (magnification, ×400). Cells were stained with Giemsa, colonies were counted. The graph represents means of three independent experiments ± standard deviations (SD)
PDK1 Is the Direct Target miR-375
Using the algorithms for target gene prediction, including PicTar, TargetScan, and miRanda, PDK1 was identified as one of the potential targets of miR-375. The predicted binding of miR-375 with PDK1 3′UTR is illustrated in Fig. 4a. To further confirm that PDK1 is the direct target of miR-375, a segment of the 3′UTR of PDK1, with or without point mutations sequence, was sub-cloned downstream of the firefly luciferase reporter. The constructs were then co-transfected with miR-375 precursor or with pre-miR control for luciferase activity assays. The relative luciferase activity of the WT construct of PDK1 3′UTR in both the esophageal cancer cells was significantly reduced in the presence of miR-375 (P < 0.05 for EC9706 cells and P < 0.01 for EC-1 cells), whereas such a suppressive effect of miR-375 on luciferase activity was not observed in both cells with the MUT construct of PDK1 3′UTR (Fig. 4b), highlighting a direct and specific interaction of miR-375 on PDK1 3′UTR.

a Predicted binding of miR-375 to PDK1 3′UTR. b The firefly luciferase reporter activity is significantly reduced in the PDK-UTR vector compared with PDK-UTR-Mut in EC9706 and EC-1 cells (P < 0.001). The data were normalized to Renilla luciferase activities. Values are expressed as the mean ± SD of three replicate experiments. c Cells were subjected to Western blot analysis using antibodies against PDK1, the phosphorylated form of Akt (Ser473), total Akt and Actin. Actin was used as an internal control. AC anti-miR-375 control
To further substantiate PDK1 as a target of miR-375, we transfected EC9706 and EC-1 cells with miR-375 precursors and anti-miR-375 followed by Western blotting for PDK1. Re-expression of miR-375 in EC9706 and EC-1 cells decreased expression of PDK1 protein (Fig. 4c). The expression of PDK1 in these esophageal cancer samples was also determined. The expression of PDK1 was detected in 83.3% (25 of 30) of cases with miR-375 promoter hypermethylated, and in 30% (3 of 10) of cases without miR-375 promoter hypermethylated (P = 0.003). Representative examples of PDK1 protein expression in esophageal cancer samples are shown in Fig. 5. Together these data indicate that PDK1 is a target for miR-375 in esophageal cancer cells.

Immunohistochemical staining for PDK1 with anti-PDK1 in the cancerous and normal tissues. The nuclei were counter-stained with hematoxylin. The scores of PDK1 expression were graded according to the number of stained cells
The phosphorylated and total Akt was also analyzed, as it has recently been reported that miR-375 regulates the PDK1/Akt survival pathway in esophageal cancer. The phosphorylation of Akt on Ser473 was markedly decreased, whereas the total Akt level was rarely altered. When the EC9706 and EC-1 cells were transfected with siRNA targeting PDK1, the percentage of apoptosis was dramatically enhanced (Fig. 6). It was similar to that caused by transfection of miR-375 in both cell lines. These findings suggests that inactivation of PDK1 is involved in miR-375-induced apoptosis.

a Western-blot analysis of EC9706 and EC-1 72 h after transfected with siPDK1 or control oligo. b The apoptotic percentage of the cells with siPDK1 molecule or control oligo was detected by flow cytometric analysis. Values are expressed as the mean ± SD
Discussion
Studies have reported that miR-375 downregulation was common in various cancers, such as pancreatic, hepatocellular, gastric, head and neck cancer [4, 8, 10, 11]. In this study, we confirmed that miR-375 was frequently downregulated in esophageal cancer tissues compared to corresponding non-cancerous tissues. Epigenetic inactivation of miRNAs in human cancer constitutes an emerging mechanism involved in the progression of cancer [12, 13]. Studies revealed that epigenetic silencing of miR-375 has been found in hepatocellular, gastric and breast cancer [8, 14]. We determined whether this downregulation is also mediated by epigenetic mechanisms in esophageal cancer. We found that the miR-375 promoter was methylated in 30 of 40 (75%) cancer tissues and 2 of 40 (5%) in adjacent non-cancerous tissues. The expression of miR-375 was further determined in a panel of five human esophageal cancer cell lines (EC9706, EC109, HCT-15, TE-1 and EC-1). We examined a further link between miR-375 CpG island hypermethylation and its gene silencing by the treatment of these cancer cell lines with DAC (a DNA demethylating agent). After the treatment of DAC, the expression of miR-375 was restored.
From a functional standpoint, we next wanted to examine whether epigenetic inactivation of miR-375 inhibited growth suppression in esophageal cancer cells. By restoring miR-375 expression in esophageal cancer cells, we indeed showed that miR-375 suppressed cell growth, induced apoptosis and inhibited the anchorage-independent growth in esophageal cancer cells, suggesting a tumour suppressive role of miR-375. The expression of PDK1 in these esophageal cancer samples was also determined. In addition, we found that the expression of PDK1 was detected in 83.3% (25 of 30) of cases with miR-375 promoter hypermethylated, and in 30% (3 of 10) of cases without miR-375 promoter hypermethylated. This indicates that the miR-375 promoter hypermethylation is significantly correlated with expression of PDK1 (P = 0.003).
Aberrant activation of the phosphatidylinositol-3 kinase (PI3 K)/Akt pathway plays a fundamental role in esophageal tumorigenesis [15, 16]. Activation of PI3 K leads to the recruitment of PDK1 to the proximal side of the plasma membrane, which results in the activation of AKT kinase and p90RSK, a kinase downstream of mitogen-activated protein kinase (MAPK) that is important for cell proliferation and survival [17–19]. In addition, loss of PDK1 sensitizes tumor cells to chemotherapeutic agents and radiation and results in significant cell growth inhibition and apoptosis, suggesting that PDK1 plays a pivotal role in MAPK and PI3 K signaling in tumor cells [19–21]. PDK1 is found to be a direct target of miR-375 in gastric cancer cells, and ectopic expression of miR-375 caused reduction of PDK1 expression. Similarly, we found that exogenous of miR-375 suppressed the expression of PDK1, resulting in decreased phosphorylation of Akt. PDK1 was shown to be a direct target of miR-375 by luciferase reporter assay. Apoptosis in esophageal cancer cells was induced when PDK1 was inactivated by siRNAs, which was to ectopic expression of miR-375. It was suggested that inactivation of the PDK1/Akt pathway is involved in miR-375-induced apoptosis in esophageal cancer.
In conclusion, miR-375 was frequently downregulated in esophageal cancer and miR-375 suppressed cell growth, induced apoptosis and inhibited the cell cycle in esophageal cancer cells. These findings may provide a potential development of miRNA-based targeted approaches for the treatment of esophageal cancer. Our results suggest that miR-375 may have a tumor suppressor function by directly targeting PDK1 to inhibit the proliferation and cell cycle of esophageal cancer cells.
References
Avissar M, Christensen BC, Kelsey KT, Marsit CJ. MicroRNA expression ratio is predictive of head and neck squamous cell carcinoma. Clin Cancer Res. 2009;15:2850–2855.
Avissar M, McClean MD, Kelsey KT, Marsit CJ. MicroRNA expression in head and neck cancer associates with alcohol consumption and survival. Carcinogenesis. 2009;30:2059–2063.
Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500.
Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963.
Wang WX, Wilfred BR, Madathil SK, et al. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am J Pathol. 2010;177:334–345.
Liu M, Lang N, Qiu M, et al. miR-137 targets Cdc42 expression, induces cell cycle G1 arrest, and inhibits invasion in colorectal cancer cells. Int J Cancer. 2011;128:1269–1279.
Zhao JJ, Lin J, Lwin T, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. 2010;115:2630–2639.
Ding L, Xu Y, Zhang W, et al. MiR-375 frequently downregulated in gastric cancer inhibits cell proliferation by targeting JAK2. Cell Res. 2010;20:784–793.
de Souza Rocha Simonini P, Breiling A, Gupta N, et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010;70:9175–9184.
Liu AM, Poon RT, Luk JM. MicroRNA-375 targets Hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem Biophys Res Commun. 2010;394:623–627.
Mathe EA, Nguyen GH, Bowman ED, et al. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: Associations with survival. Clin Cancer Res. 2009;15:6192–6200.
Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–776.
El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, van Obberghen E. miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes. 2008;57:2708–2717.
Tsukamoto Y, Nakada C, Noguchi T, et al. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res. 2010;70:2339–2349.
Wei L, Xu Z. Cross-signaling among phosphinositide-3 kinase, mitogen-activated protein kinase and sonic hedgehog pathways exists in esophageal cancer. Int J Cancer. Epub. 2010/09/13.
Li B, Tsao SW, Li YY, et al. Id-1 promotes tumorigenicity and metastasis of human esophageal cancer cells through activation of PI3K/AKT signaling pathway. Int J Cancer. 2009;125:2576–2585.
Lu Z, Cox-Hipkin MA, Windsor WT, Boyapati A. 3-phosphoinositide-dependent protein kinase-1 regulates proliferation and survival of cancer cells with an activated mitogen-activated protein kinase pathway. Mol Cancer Res. 2010;8:421–432.
Rodriguez OC, Lai EW, Vissapragada S, et al. A reduction in PTEN tumor suppressor activity promotes ErbB-2-induced mouse prostate adenocarcinoma formation through the activation of signaling cascades downstream of PDK1. Am J Pathol. 2009;174:2051–2060.
Liu Y, Wang J, Wu M, et al. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Mol Cancer Res. 2009;7:944–954.
Ballif BA, Shimamura A, Pae E, Blenis J. Disruption of 3-phosphoinositide-dependent kinase 1 (PDK1) signaling by the anti-tumorigenic and anti-proliferative agent n-alpha-tosyl-l-phenylalanyl chloromethyl ketone. J Biol Chem. 2001;276:12466–12475.
Finlay DK, Sinclair LV, Feijoo C, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med. 2009;206:2441–2454.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, X., Lin, R. & Li, J. Epigenetic Silencing of MicroRNA-375 Regulates PDK1 Expression in Esophageal Cancer. Dig Dis Sci 56, 2849–2856 (2011). https://doi.org/10.1007/s10620-011-1711-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-011-1711-1