
Abstract
Introduction
MicroRNAs (miRNAs) are a class of small (19–25 nucleotides) noncoding RNAs that regulate the expressions of a wide variety of genes, including some involved in cancer development. Some recent studies show that DNA methylation contributes to down-regulation of microRNA-137 (miR-137) during tumorigenesis. Whether down-regulation of miR-137 also exists in gastric cancer is unknown.
Aim
Our aim was to test the hypothesis that down-regulation of miR-137 also exists in gastric cancer.
Methods
Expression of levels of miR-137 were examined using real-time PCR on paired gastric cancer and adjacent non-cancerous tissues. The methylation status is detected by MSP.
Results
Results show that miR-137 is downregulated by hypermethylation of the promoter in gastric cancer tissues. Epigenetic silencing of miR-137 induced an up-regulation of its targets, Cdc42. Restoration of the miR-137 expression in gastric cancer cell lines downregulated the Cdc42 expression. Restoration of the miR-137 and inactivation of Cdc42 induce apoptosis and cell cycle G1 arrest in gastric cancer cells. Furthermore, the miR-137 expression was found to be inversely correlated with CDC42 expression in gastric caner.
Conclusions
miR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs are 19–25-nucleotide regulatory non-proteincoding RNA molecules that usually function as endogenous repressors of target genes, resulting in mRNA degradation or inhibition of translation [1, 2]. The downregulation of miRNA subsets implies a tumor-suppressor function, which is often observed in tumor development [3, 4]. miR-137 has been found to be downregulated in colorectal cancer glioblastoma multiforme, oral cancer, and squamous cell carcinoma of the head and neck (SCCHN) [5, 6]. Cdc42 and micropthalmia-associated transcription factor (MITF) are all found to be the target of miR-137 [7, 8]. Methylation of the miR-137 CpG island was a cancer-specific event and was frequently observed in colorectal cancer (CRC) and oral cancer [9, 10].
Cdc42 (cell division cycle 42), is a small GTPase of the Rho-subfamily. It maps to 1p36.1 and encodes a 25-kDa protein [11]. The family of small Rho GTPases act as molecular switches that control key signaling of the cell in response to diverse extracellular stimuli. The Cdc42 gene and has been implicated in cancer initiation and progression [12]. Cdc42 can interact with different effector molecules which, in turn, regulate actin cytoskeleton, microtubule networks, cell polarity, proliferation, apoptosis, endocytosis, and secretion. High levels of Cdc42 expression have been reported in several cancers, suggesting a role of Cdc42 in cancer [13–15].
Studies have extensively shown that hypermethylation of the gene promoter is a frequent mechanism of silencing and a finding associated with the prognosis of the diseases and response to therapy in patients with cancer. In the present study, we have identified that miR-137 is downregulated by hypermethylation of the promoter in gastric cancer tissues. Epigenetic silencing of miR-137 induced an up-regulation of its target, Cdc42. Restoration of the miR-137 expression in gastric cancer cell lines downregulated the Cdc42 expression. Furthermore, we found that the inhibition of cell cycle progression and induction of apoptosis after re-expression of miR-137 was partially through suppression of Cdc42.
Materials and Methods
Patients
Human gastric cancer samples were collected from surgical specimens from 40 patients with gastric cancer at Department of Medicine-Oncology, the First Affiliated Hospital, Zhengzhou University, Zhengzhou, China. Non-tumor samples from the macroscopic tumor margin were isolated at the same time and used as the matched adjacent non-neoplastic tissues (>5 cm). All the samples were divided into two parts. Tissue samples were collected, immediately snap frozen in liquid nitrogen, and stored at −80°C until RNA extraction. All samples were obtained with informed consent and with institutional review board approval of the hospital. All patients obtained a confirmed diagnosis of gastric carcinoma after resection. Five gastric cancer cell lines (AGS, SGC-7901, MKN28, MKN45, and BCG823) were all preserved in our laboratory and maintained in RPMI 1640 with 10% FBS.
Real-Time qPCR
Total RNA was extracted from gastric cancer or normal tissues using TRIZOL (Invitrogen), according to the manufacturer’s instructions. For miRNA qPCR, reverse transcription was performed using the QuantMir RT KIT (System Bioscience). In brief, 1 μg of RNA containing miRNA was polyadenylated by poly(A) polymerase and then reverse transcribed to cDNA using oligo-dT primers. The cDNA then serves as the template for SYBR real-time PCR using SYBR-green PCR Master Mix (Applied Biosystems). The miR-137-specific forward primer sequence was designed on the basis of miRNA sequences obtained from the miRBase database. Human U6 snRNA was used for normalization. For Cdc42 mRNA qPCR, reverse transcription was carried out using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, Promega). Following the protocol of the manufacturer, the amount of Cdc42 expression, normalized to a human actin endogenous reference, is given by: 2−∆Ct. Real-time RT-PCR was repeated at least three times for each specimen, and the mean was obtained.
DNA Methylation Analysis of the miR-137 Gene
Genomic DNA (2 μg) was modified with sodium bisulfite using EpiTect Bisulfite kit (Qiagen). Methylation status was analyzed by bisulfite genomic sequencing of the CpG islands. The fragment covering 14 CpG sites from miR-137 promoter region was amplified from bisulfite-modified DNA. The primers used are shown in Fig. 1a. Amplified bisulfite-sequencing PCR products were cloned into pMD18-T simple vector (Takara). Methylation status of human gastric normal tissues and tumor samples was examined by methylation-specific PCR (MSP) analysis. We investigated the effect of a demethylating agent, 5′-aza-2′-deoxycytidine (DAC, Sigma) on the expression of miR-137 in AGS, MKN28, SGC-7901, BCG823, and MKN45 cells. Cells were plated at a density of 2 × 105 per well in six-well plates for 18 h, then treated with DAC at concentrations of 10 μM/l in duplicate. After treatment for 48 h, the cells were harvested.

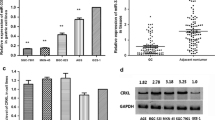
a The relative miR-137 expression between tumor tissues and their paired adjacent non-cancerous tissues from 40 gastric cancer patients using real-time qPCR. Expression of miR-137 (Log10 scale at Y axis) was normalized to U6. Statistical difference was analyzed using Wilcoxon’s test, p < 0.01. b Representative MSP results of miR-137 hypermethylation in gastric cancer tissues. Case numbers are shown on top. M methylated primers; U unmethylated primers. c Relative miR-137 expression in 40 gastric cancer tissues stratified by miR-137 promoter methylation. miR-137 levels and promoter methylation were determined by qRT-PCR and MSP, respectively. Depending on each tumor’s promoter methylation status, the cases were subdivided into two groups, i.e., unmethylated and methylated. Note significantly lower mean miR-137 expression in the tumors with promoter methylation (p < 0.01, two-sided t test)
Cell Transfection
The pre-miR-137 precursor molecule (miR-137 mimics) and negative control RNA-oligonucleotides were gained from Ambion corporation (Ambion, Austin, USA). The day before transfection, gastric cancer cells were seeded in antibiotic-free medium. Transfection of miRNAs was carried out using Lipofectamine 2000 in accordance with the manufacturer’s procedure (Invitrogen, CA, USA). The level of miR-137 mimics expression in the gastric cancer cells was assayed by real-time PCR.
Cell Growth Assay
Gastric cancer cells were seeded in 96-well plates 1 day before transfection. After transfecting with miR-137, control oligo, the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) was used to determine relative cell growth.
Cell Cycle Assay
Cells were harvested by trypsinization 72 h after transfection, washed three times with ice-cold PBS, and fixed with 70% ethanol overnight at 4°C. The fixed cells were rehydrated in PBS and subjected to PI/RNase staining followed by fluorescence-activated cell sorter scan (FACS) analysis (Becton Dickinson, Mountain View, CA, USA). The percentage of cells in each phase of the cell cycle was estimated using ELITE software.
Evaluation of Apoptosis
Apoptosis was detected by flow cytometric analysis of Annexin V staining. Annexin V–FITC versus PI assay was performed as previously reported. Briefly, adherent cells were harvested and suspended in the Annexin-binding buffer (1 × 106 cells/ml). Thereafter, cells were incubated with Annexin V–FITC and PI for 15 min at room temperature in the dark and immediately analyzed by flow cytometry. The data are presented as bi-parametric dot plots showing Annexin V–FITC green fluorescence versus PI red fluorescence.
Luciferase Activity Assay
A fragment of the wild-type (WT) Cdc42 3′UTR containing the predicted miR-137 binding site was amplified by RT-PCR. Site-directed mutagenesis of the miR-137 target site was carried out using the Stratagene Quik-Change site-directed mutagenesis kit (Stratagene, Heidelberg, Germany). The construct was sequenced and named Cdc-UTR-Mut. The pMIR-report luciferase vector was used for the construction of Cdc-UTR or Cdc-UTR-Mut plasmids (Ambion, USA). AGS and MKN45 cells were cultured in 24-well plates. In each well, 10 ng of phRL-TK Renilla luciferase vector (Promega, USA) was co-transfected to normalize transfection efficiency, and 500 ng of Cdc-UTR or Cdc-UTR-Mut plasmids together with 10 nM miR-137 mimics or negative control was also co-transfected. Transfection was done using Lipofectamine 2000 and Opti-MEM I reduced-serum medium (Life Technologies, CA, USA). Firefly luciferase activity was measured using the Dual Luciferase Assay kit (Promega). Normalized relative luciferase activity (RLA) was calculated as the following formula:
Western-Blot Analysis
Protein of treated cell lines was extracted by mammalian protein extraction reagent (Pierce, USA), supplemented with protease inhibitors cocktail (Sigma, USA). Protein samples (50 μg) were resolved by 10% SDS-PAGE and then transferred to PVDF membranes. Autoradiograms were quantified by densitometry (Quantity One software; Bio-Rad, Hercules, CA). Actin-specific antibody was used for loading control. Mouse monoclonal anti-Cdc42 (1:1000, Cell Signaling Technology), anti-phosphorylated ERK1/2 antibody, total ERK1/2 (1:1000, Cell Signaling Technology) and Mouse monoclonal anti-Actin (1:1000, abcam, USA) were used.
Statistical Analysis
Expression of miR-137 in paired gastric cancer and adjacent non-cancerous tissues were compared by paired t test. The difference between the two groups in cell proliferation assay and luciferase reporter assay was analyzed by two-sided Student’s t test. Data are expressed as the mean ± SD from at least three independent experiments. All p values are two-sided and a value of <0.05 was considered to be statistically significant.
Results
Validation of Downregulated miR-137 in Gastric Cancer
First, we quantified the expression of miR-137 in gastric cancer tissues by using qRT-PCR. Our results showed that miR-137 level was significantly decreased in 33 of 40 (82.5%) gastric cancer tissues when compared to that in their adjacent normal tissues (p < 0.01; Wilcoxon’s paired test; Fig. 1a).
Involvement of DNA Methylation in Downregulation of miR-137
It was reported that the expression of miR-137 was regulated by CpG island methylation in colorectal cancer. Next, we determined whether this downregulation is also mediated by epigenetic mechanisms in gastric cancer. The methylation of promoter was detected by methylation-specific PCR (MSP). We found that the miR-137 promoter was methylated in 28 of 40 (70%) cancer tissues and four of 40 (10%) in adjacent non-cancerous tissues (p < 0.01). Representative examples of the gel analysis of MSP are shown in Fig. 1b. Correlation of the promoter methylation data with miR-137 expression revealed a significantly lower mean expression level in the 28 tumors with promoter methylation as compared to the tumors without promoter methylation (Fig. 1c).
The methylation of miR-137 was further determined in a panel of five human gastric cancer cell lines (AGS, SGC-7901, MKN28, MKN45, and BCG823). The area of the CpG-rich region around the transcription initiation site of miR-137 gene which spanned 14 CpG sites was sequenced (Fig. 2a). As shown in Fig. 2b, most CpG dinucleotides were methylated in gastric cancer cell lines. To confirm that this downregulation was because of the miR-137 promoter methylation, gastric cancer cell lines were treated with DAC for 48 h and RT-PCR was performed to detect miR-137 expression (Fig. 3a). The demethylation of miR-137 by DAC in these gastric cancer cells was verified by the use of BSP (Fig. 3b). After treatment with DAC, we found that the levels of miR-137 were induced in these gastric cancer cell lines, suggesting that the miR-137 gene silencing is accounted for by hypermethylation.

a A map of the CpG islands in relation to the promoter of the miR-137. The locations of sense and antisense primers used for bisulfite-sequencing PCR are indicated by brackets. The CpG sites are underlined. b Methylation patterns of individual bisulfite-sequenced clones of the miR-137 promoter in gastric cancer cell lines. Black and white areas represent, respectively, the percentage of methylated and unmethylated CpG sites out of the colonies sequenced for each case

a The expression of miR-137 in gastric cancer cell lines treated with or without demethylation agent DAC as determined by RT-PCR. b An illustrative fragment of the sequencing electropherogram is shown for AGS cells treated with or without DAC
Ectopic miR-137 Expression Alters Gastric Cancer Cell Growth, Apoptosis, and Cell Cycle
Frequent downregulation of miR-137 in gastric cancer cell lines and primary gastric cancer tissues implies that miR-137 may have a role in gastric cancer carcinogenesis. To prove this, the effect of ectopic expression of miR-137 on cell growth was investigated in two gastric cancer cell lines (AGS and MKN45). We transfected 50 nM of miR-137 RNA oligonucleotides (Ambion) into AGS and MKN45 cells followed by analysis using CCK-8 assay 48 h later. Scrambled oligonucleotides purchased from Ambion were used as a negative control. All transfections and CCK-8 assays were performed in triplicate in 96-well plates. As shown in Fig. 4a, the increased expression of miR-137 by ectopic miR-137 expression significantly inhibited the growth of AGS and MKN45 cells (all p values < 0.05; Mann–Whitney test). To further understand the mechanisms by which cell proliferation is affected, flow cytometry was performed to analyze the cell cycle phase distribution of these cells. The cell-cycle progression of AGS and MKN45 cells transfected with miR-137 was stalled at the G1 phase with a significant decrease in S and G2/M phases compared to cells transfected with control oligo (Fig. 4b). We sought to evaluate whether the effect of cell growth was related to apoptosis; 30 ± 4% of AGS and 25 ± 3% of MKN45 cells were stained positively with Annexin V as determined by FCM, whereas there were no significant changes in the control cells (Fig. 4c) (p < 0.05).

a Cellular proliferation was measured by CCK-8 assay in gastric cancer cells transfected with miR-137 mimics or control (Mock). Points represent means of three independent experiments ± standard deviations. b miR-137 induces cell-cycle G1 arrest in gastric cancer cells. Cell-cycle progression of AGS and MKN45 cells 72 h after being transfected with miR-137 or negative control (Mock) was determined by FACS analysis. Data are representative of three independent experiments. Asterisk indicates a significant difference from control oligo-transfected control cells (p < 0.05). c The apoptotic percentage of the cells with miR-137 precursor molecule or Mock was detected by flow cytometric analysis. The values above each bar represent the fraction of Annexin V+/PI− and annexin V+/PI+. Values are expressed as the mean ± SD of three replicate experiments. *p < 0.001 compared to Mock
Cdc42 Is the Direct Target miR-137
Using the algorithms for target gene prediction, including PicTar, TargetScan, and miRanda, Cdc42 was identified as one of the potential targets of miR-137. To further confirm that Cdc42 is the direct target of miR-137, a segment of the 3′UTR of Cdc42, with or without point mutations sequence, was sub-cloned downstream of the firefly luciferase reporter. The constructs were then co-transfected with miR-137 precursor or with pre-miR control for luciferase activity assays. The relative luciferase activity of the WT construct of Cdc42 3′UTR in both the gastric cancer cells was significantly reduced in the presence of miR-137 (p < 0.05), whereas such a suppressive effect of miR-137 on luciferase activity was not observed in both cells with the MUT construct of Cdc42 3′UTR (Fig. 5a), highlighting a direct and specific interaction of miR-137 on Cdc42 3′UTR.

a The firefly luciferase reporter activity is significantly reduced in Cdc-UTR vector compared to Cdc-UTR-Mut in AGS and MKN45 cells (p < 0.001). The data were normalized to Renilla luciferase activities. Values are expressed as the mean ± SD of three replicate experiments. b Cells were subjected to Western-blot analysis using antibodies against Cdc42, the phosphorylated form of ERK1/2, total ERK1/2, and Actin. Actin was used as an internal control. c The apoptotic percentage of the cells with siRNA or Mock was detected by flow cytometric analysis. d Comparison of miR-137 expression (x axis) to Cdc42 mRNA expression (y axis) in the gastric cancer samples. Inverse correlation was also obtained by Spearman’s correlation, r < −0.72, p < 0.01
To further substantiate Cdc42 as a target of miR-137, we transfected AGS and MKN45 cells with miR-137 precursors or siRNA targeting Cdc42 followed by Western blotting for Cdc42. Re-expression of miR-137 in AGS cells decreased expression of Cdc42 protein. Similar results were obtained when cells were transfected with siRNA targeting Cdc42 (Fig. 5b). The results were also observed in MKN45 cells (Supplementary Fig. 1). Together, these data indicate that Cdc42 is a target for miR-137 in AGS cells. The Cyclin D1, phosphorylated and total ERK1/2 was also analyzed, as it has recently been reported that miR-137 regulates the Cdc42/ERK signaling in colorectal cancer. The Cyclin D1 and phosphorylation of ERK1/2 were markedly decreased, whereas the total ERK1/2 level was rarely altered. When the AGS cells were transfected with siRNA targeting Cdc42, the percentage of apoptosis was dramatically enhanced (Fig. 5c). It was similar to that caused by transfection of miR-137 in this cell line. After treatment with DAC, we also found that the level of Cdc42 was reduced in MKN45 cells (Supplementary Fig. 2), suggesting that miR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42. These findings also suggest that inactivation of Cdc42 is involved in miR-137-induced apoptosis.
To confirm the relevance of the expression of Cdc42 and the relationship between miR-137 and Cdc42, we assessed the expressions of miR-137 and Cdc42 mRNA in an independent set of human gastric cancer tissues and their adjacent non-cancerous tissues from 15 patients. We showed that expressions between miR-137 and Cdc42 mRNA were inversely correlated in all 15 paired gastric cancer and adjacent non-cancerous tissues (r = −0.72, p < 0.01; Spearman’s correlation; Fig. 5d).
Discussion
miRNAs are 16-to 25-nucleotide-long RNAs, able to bind complementary sequences in 3-untranslated regions (3′UTR) of several target mRNAs to induce their degradation or translational repression. They are deemed to play a crucial role in the initiation and progression of human cancer. Epigenetic inactivation of miRNAs in human cancer constitutes an emerging mechanism involved in the progression of cancer. Studies have reported that miR-137 downregulation was common in various cancers, such as pancreatic, hepatocellular, and head and neck cancers.
Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Expression of miR-137 was restricted to the colonocytes in normal mucosa and inversely correlated with the level of methylation. The promoter methylation of miR-137 was associated with female gender and inversely associated with body mass index (BMI) in squamous cell carcinoma of the head and neck (SCCHN). Promoter methylation of miR-137 appears to be a relatively frequently detected event in oral rinse of SCCHN patients and may have future utility as a biomarker in DNA methylation panels [10]. In this study, we confirmed that miR-137 was frequently downregulated in gastric cancer tissues than in their corresponding non-cancerous tissues.
We determined whether downregulation of miR-137 is also mediated by epigenetic mechanisms in gastric cancer. We found that the miR-137 promoter was methylated in 21 of 30 (70%) cancer tissues and two of 30 (6.7%) in adjacent non-cancerous tissues. The expression of miR-137 was further determined in a panel of four human gastric cancer cell lines (AGS, SGC-7901, MKN28, MKN45, and BCG823). We examined a further link between miR-137 CpG island hypermethylation and its gene silencing by the treatment of these cancer cell lines with DAC. After the treatment of DAC, the expression of miR-137 was restored.
From a functional standpoint, we next wanted to examine whether epigenetic inactivation of miR-137 inhibited growth suppression in gastric cancer cells. By restoring miR-137 expression in gastric cancer cells, we indeed showed that miR-137 suppressed cell growth, induced apoptosis and inhibited the cell cycle in gastric cancer cells, suggesting a tumor-suppressive role of miR-137.
Three independent studies screening for important oncogenes have found that Cdc42 is commonly altered by retroviral insertions. Cdc42 was found to be overexpressed in colorectal cancer samples, and this expression was associated with silencing of ID4 with statistical significance [13]. The up-regulated Cdc42 activity may impair c-Cbl-mediated EGFR degradation, contribute to EGFR hyperactivity, and induce proteasomal degradation of p21CIP1, leading to an increase in cell proliferation and migration. These functional outcomes may be through regulation of PAK1, GSK3b, MLC, ERK1/2, and JNK pathways. In addition, downregulation of Cdc42 signals can inhibit anchorage-independent growth and induce apoptosis via the PI(3)K-Akt and Erk signaling cascades and the p53 tumor suppressor [16, 17].
Cdc42 is found to be a direct target of miR-137 in colorectal cancer cells, and ectopic expression of miR-137 caused reduction of Cdc42 expression [8]. Similarly, we found that exogenous of miR-137 suppressed the expression of Cdc42, resulting in decreased phosphorylation of ERK1/2. Similar results were also obtained when siRNAs targeting Cdc42 was transfected. CDC42 was shown to be a direct target of miR-137 by luciferase reporter assay. Additionally, we found that inactivation of Cdc42 by siRNA and the exogenous expression of miR-137 induce apoptosis in AGS cells. Apoptosis in gastric cancer cell induced by siRNAs targeting Cdc42 was similar to the values obtained from ectopic expression of miR-137. It was suggested that inactivation of the Cdc42/ERK pathway is involved in miR-137-induced apoptosis in gastric cancer.
Taken together, our data indicate that miR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42. Exogenous expression of miR-137 and Cdc42 siRNA can effectively down-regulate Cdc42 overexpression and induce apoptosis in gastric cancer, suggesting that the combined miRNA-based and epigenetic treatment may be a novel potential therapeutic target for human gastric cancer.
References
Zhang B, Pan X, Cobb GP, Anderson TA. MicroRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12.
Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005;15:563–568.
Huang YW, Liu JC, Deatherage DE, et al. Epigenetic repression of microRNA-129–2 leads to overexpression of sox4 oncogene in endometrial cancer. Cancer Res. 2009;69:9038–9046.
Dyrskjot L, Ostenfeld MS, Bramsen JB, et al. Genomic profiling of microRNAs in bladder cancer: Mir-129 is associated with poor outcome and promotes cell death in vitro. Cancer Res. 2009;69:4851–4860.
Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–2105.
Silber J, Lim DA, Petritsch C, et al. Mir-124 and mir-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14.
Bemis LT, Chen R, Amato CM, et al. Microrna-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer Res. 2008;68:1362–1368.
Liu M, Lang N, Qiu M, Xu F, Li Q, Tang Q, Chen J, Chen X, Zhang S, Liu Z, Zhou J, Zhu Y, Deng Y, Zheng Y, Bi F. Mir-137 targets cdc42 expression, induces cell cycle g1 arrest, and inhibits invasion in colorectal cancer cells. Accepted in Int J Cancer.
Balaguer F, Link A, Lozano JJ, Cuatrecasas M, Nagasaka T, Boland CR, Goel A. Epigenetic silencing of mir-137 is an early event in colorectal carcinogenesis. Cancer Res. 2010;70:6609–6618.
Langevin SM, Stone RA, Bunker CH, Grandis JR, Sobol RW, Taioli E. Microrna-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis. 2010;31:864–870.
Szczur K, Zheng Y, Filippi MD. The small Rho GTPase Cdc42 regulates neutrophil polarity via CD11b integrin signaling. Blood. 2009;114:4527–4537.
Feng Y, Hartig SM, Bechill JE, Blanchard EG, Caudell E, Corey SJ. The cdc42-interacting protein-4 (cip4) gene knock-out mouse reveals delayed and decreased endocytosis. J Biol Chem. 2010;285:4348–4354.
Gomez Del Pulgar T, Valdes-Mora F, Bandres E, et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int J Oncol. 2008;33:185–193.
Sahai E, Marshall CJ. Rho-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142.
Kamai T, Yamanishi T, Shirataki H, et al. Overexpression of RhoA, Rac1, and Cdc42 GTPases is associated with progression in testicular cancer. Clin Cancer Res. 2004;10:4799–4805.
Yang L, Wang L, Zheng Y. Gene targeting of Cdc42 and Cdc42GAP affirms the critical involvement of Cdc42 in filopodia induction, directed migration, and proliferation in primary mouse embryonic fibroblasts. Mol Biol Cell. 2006;17:4675–4685.
Zugasti O, Rul W, Roux P, et al. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and Cdc42 via Akt. Mol Cell Biol. 2001;21:6706–6717.
Conflict of interest
The author(s) declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Qingjiang Chen and Xiaobing Chen contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.

10620_2010_1536_MOESM1_ESM.jpg
{kind=link}
MKN45 cells were subjected to Western blot analysis using antibodies against Cdc42, the phosphorylated form of ERK1/2, total ERK1/2 and Actin. Actin was used as an internal control. (JPEG 33 kb)

10620_2010_1536_MOESM2_ESM.jpg
{kind=link}
The expression of Cdc-42 in gastric cancer cell lines MKN45 treated with or without demethylation agent DAC as determined by Western blot. Actin was used as an internal control. (JPEG 74 kb)
Rights and permissions
About this article
Cite this article
Chen, Q., Chen, X., Zhang, M. et al. miR-137 Is Frequently Down-Regulated in Gastric Cancer and Is a Negative Regulator of Cdc42. Dig Dis Sci 56, 2009–2016 (2011). https://doi.org/10.1007/s10620-010-1536-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-010-1536-3