
Abstract
Background
Chronic inflammation of mucosal surfaces is an aberrant immune response to luminal bacteria and generates an array of oxygen radicals leading to tissue destruction and loss of function, as noted in IBD and periodontitis. We hypothesized that mucosal injury after “oral delivery” of dextran sulfate sodium (DSS) or TNBS for an extended period of 18 weeks is reflected by chronic inflammatory responses in a time-dependent fashion.
Methods
Dextran sulfate sodium was administered in the diet biweekly; TNBS or sham controls was administered orally twice a week. Additional groups received TNBS or sham injections into gingival tissue.
Results
Animals tolerated oral applications with no severe clinical symptoms. The DSS-group developed diarrhea during the period of administration, and returned to normal during DSS abstinence. The TNBS-group developed no systemic clinical symptoms. Splenic length and weight increased in the DSS-group in a time-dependent fashion (P < 0.01) and remained normal in the TNBS-group. Colons from the DSS-group were significantly shortened (P < 0.001) and colonic weight increased compared with controls or the TNBS-group (P < 0.05). The DSS-group developed extensive dilation of the stomach wall, ileum, and megacolon, with abdominal fat deposits. In addition, the DSS-group showed dysregulated hepatic concentrations of antioxidants (i.e. cysteine, GSH, SAMe) in a time-dependent manner that correlated with a significance increase in alveolar bone resorption. Localized TNBS-mucosal delivery caused severe inflammation, granuloma formation, and rapid bone resorption.
Conclusions
This model of mucosal stimulation eliciting chronic inflammatory responses in the gut and oral cavity mimics aspects of IBD and periodontal disease progression in patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic inflammation of mucosal surfaces generally is an aberrant immune response by the mucosal tissues to luminal bacteria. This response generates an array of oxygen radicals leading to inflammation, soft and hard tissue destruction, and loss of function, as noted in inflammatory bowel disease [1–3] and periodontitis [4–9]. In both IBD and periodontitis, PMNs release destructive ROS, e.g. superoxide, via the respiratory burst [4–6], proteinases, and other mediators that can damage host tissues [7, 10–12]. These molecules induce oxidative damage to mucosal tissue and in the oral cavity lead to osteoclastic bone resorption [7, 13–15]. IBD results in the production of both inflammatory mediators and high levels of ROS in the local microenvironment of the gut inflammation. The oral sequelae in IBD, especially Crohn’s disease, are well-established [16–22]. In addition, saliva of Crohn’s disease patients exhibits abnormal oxidative stress, NO, and TGF-β, which underlines the importance of oxidative stress in the pathogenesis of Crohn’ disease [22].
About one half of the world’s population suffer from periodontitis or lack of teeth by the age of 50 years [23]. The chronic inflammation of periodontitis is initiated by complex pathogenic subgingival biofilms containing several likely periodontal pathogens, generally regarded to be Gram-negative anaerobic commensal microbiota which are opportunistic pathogens in the disease [10].
Although oral mucosal and periodontal disease occur in patients suffering from IBD, it is not clear whether periodontal disease is a common manifestation or a sporadic event in IBD patients. Additionally, some drugs used in IBD treatment can have adverse effects on any part of the gastrointestinal tract, including the oral cavity. For instance, gum hyperplasia is induced by immunosuppressant agents (i.e. cyclosporine) in IBD patients, particularly in those with poor oral hygiene [16]. There are conflicting clinical studies regarding IBD and periodontal complications. In a recent clinical study, patients with Crohn’s disease, patients with ulcerative colitis, and healthy controls were compared [17]. Significantly more IBD patients had periodontitis than controls. However, there remains minimal understanding of mechanistic relationships between IBD and gingival inflammation and destruction.
Murine models of IBD often utilize chemical induction of acute and chronic inflammation in the gut to evaluate the mechanisms of disease progression. Administration of dextran sulfate sodium (DSS) in water for 3–5 cycles induces chronic colitis similar to ulcerative colitis [3, 24–26]. Rectal exposure to trinitrobenzene sulfonic acid (TNBS) also causes an inflammatory immune response similar to the autoimmune presentation of Crohn’s disease [3, 27, 28]. Indeed, TNBS and DSS-induced inflammatory diseases utilize the murine natural microbiota and implement a reproducible model of a chronic inflammatory lesion of gut mucosal surfaces [3, 28, 29]. Various rodent models have also been used to evaluate the clinical, microbiological, and immunological aspects of periodontitis mimicking features of human disease [5, 13]. Substantial evidence in IBD patients and models [1–3] and more limited data in periodontitis [4–7] suggest similarities in the underlying inflammatory mechanisms resulting in these mucosal diseases. However, the roles of specific mediators or reactive intermediates in triggering/regulating molecular aspects of the inflammatory and innate immune responses at these mucosal sites remain to be determined.
The objectives of this study were to examine the time course of mucosal injury, after “oral delivery” of DSS and TNBS for an extended period of 18 weeks, reflected by GI and oral chronic inflammatory responses. We describe a means of inducing the chronic, progressive inflammation of IBD and oral tissues modeling periodontitis, and demonstrate broader induction of oral mucosal inflammation and disease triggered by the compounds.
Materials and Methods
Animals
Sixty-seven BALB/c mice (11–12 weeks old; Harlan Laboratories, Indianapolis, IN, USA) were housed in micro-filter top cages in an American Association of Accreditation of Laboratory Animal Care (AAALAC)-certified Laboratory Animal Research Resource Facility at the University of Kentucky Medical Center. They were placed in a room maintained at 22°C with a 12-h–12-h light–dark cycle and fed rodent chow and water ad libitum. This experimental study was approved by, and performed in accordance with the guidelines of, the Institutional Animal Care and Use Committee (IACUC). To minimize contamination, all cages were opened in a biosafety hood only.
Mice were weighed weekly and received fresh food and water three times per week. At the end of each time point, one week after the last administration of DSS or TNBS, mice were weighed and euthanized to collect samples. Finally, alveolar bone loss was determined on jaws from all animals at each time point.
Experimental Procedure
After 1 week of acclimation, on day 0 of the experiment, the BALB/c mice were weighed and ear-punched for appropriate identification and randomly assigned to each treatment group. The mice were monitored daily for comfort, food and water intake, clinical symptoms (diarrhea, rectal bleeding and pain), and survival.
TNBS Model
One group of animals was treated with trinitrobenzene sulfonic acid (TNBS, 2.5 mg solution; Sigma–Aldrich, St Louis. MO, USA) delivered via a micropipette tip (100 μl) orally twice a week.
DSS Model
Mice were treated biweekly with dextran sodium sulfate (2% DSS, American International Chemical, Framingham, MA, USA) in the diet food, followed by one week of abstinence, for a period of 7–18 weeks.
TNBS Injection
In order to explore the direct effect of local TNBS delivery into the oral gingival tissue, 50 μl TNBS was inoculated via a 28-gauge hypodermic needle into the right side of the maxillary gingiva, and the left side was injected with a vehicle control. Animals were monitored for acute inflammatory reactions and granuloma formation for 48–72 h.
Animals received fresh food and water three times a week and were weighed weekly. Control animals received a normal diet and sham challenge. At each time point five animals were euthanized [24–26, 28].
Evaluation of Colitis
The progression of colitis was evaluated over the period of 18 weeks for DSS or TNBS treatment. Physical appearance, food consumption, consistency of feces, diarrhea, presence of gross blood in stool, and body weight were monitored daily. At each time point (weeks 0, 7, 12, and 18) following initiation of the experiment the animals were overdose-anesthetized by halothane inhalation and tissue dissected. The colonic tissue was excised, perfused with phosphate-buffered saline (PBS) pH 7.4 (Sigma, St Louis, MO, USA), and weighed. A small cuff of the proximal and distal colon (within 1 cm of the rectum) was cut and fixed in 10% buffered formalin in PBS (Sigma) and processed for histopathological analysis.
The splenic and colonic tissues were weighed and measured lengthwise and a segment from each was then flash frozen in liquid nitrogen and stored at −80°C for further analysis.
The severity of the injury was determined by measurement of tissue wet length and weight [30], bowel wall thickness, and histological scores. This enabled determination of a relationship for the severity of clinical features at both mucosal sites (oral cavity, gut). A segment of liver and splenic tissue were flash frozen in liquid nitrogen and kept at −80°C for further analysis and another portion was fixed in formalin. The sections fixed in formalin were stained with hematoxylin and eosin (H&E) and evaluated by light microscopy for the presence of lesions. The severity of colitis was assessed by histological grading score of the colon. The scores were based on histological features with a numeric value (0–4) assigned to the specimen on the basis of the following criteria:
-
Grade 0
No detectable lesions, no inflammatory cells, mucosa appears normal;
-
Grade 1
Few focal inflammatory infiltrates in the mucosa;
-
Grade 2
Mild multi-focal inflammation with moderate expansion of the mucosa; crypt epithelium appears normal;
-
Grade 3
Moderate multi-focal inflammation with moderate expansion of the mucosa, mild crypt epithelium disruption;
-
Grade 4
Severe diffuse inflammation with crypt epithelium disruption and ulceration.
Whole Blood and Plasma Isolation
Immediately after euthanasia, blood was collected from the right ventricle of the heart into a syringe containing a minute amount of heparin and placed on ice. Plasma was separated by centrifugation at 5,000xg for 5 min at 4°C. Samples were kept at −80°C until further analysis.
Tissue Preparation for Antioxidant Determination
Tissue homogenates (10%, w/v) were prepared in 5% metaphosphoric acid, using all-glass Tenbroeck homogenizers, and kept on ice. After standing for 20–40 min, the homogenates were centrifuged for 1 min (10,000g) and the acid-soluble fractions were collected for measurement of free thiols/disulfides. GSH, GSSG, cysteine, and cystine were simultaneously quantified by high-performance liquid chromatography with dual electrochemical detection (HPLC–DEC). Samples (20 μl) were injected on to a 250 × 4.6 mm, 5-μm particle, C18 column (Val-U-Pak HP, fully end-capped ODS; Chrom Tech, Apple Valley, MN, USA). The injected samples were eluted isocratically with a mobile phase consisting of 0.1 M monochloroacetic acid, 2 mM heptane sulfonic acid (ion-pairing reagent), and 2% acetonitrile at pH 2.8 and delivered at a flow rate of 1 ml/min. The compounds were detected in the eluent with a Bioanalytical Systems model LC4B dual electrochemical detector, using two Au-Hg electrodes in series with potentials of −1.2 and 0.15 V for the upstream and downstream electrodes, respectively. Current (nA) was measured at the downstream electrode. Analytes were quantified from peak area measurements using authentic external standards.
Intracellular S-Adenosylmethionine
Deproteinized tissue extracts (4% metaphosphoric acid) and blood was prepared, and S-adenosylmethionine (SAMe) was determined by an HPLC method, using a 5-μm Hypersil C18 column (250 × 4.6 mm). The mobile phase consisted of 40 mM ammonium phosphate, 8 mM heptane sulfonic acid (ion-pairing reagent, pH 5.0), and 6% acetonitrile, and was delivered at a flow rate of 1.0 ml/min. SAMe was detected using a Waters 740 UV detector at 254 nm. An internal standard, S-adenosylethionine (SAE), was added to all samples and standard solutions to a concentration of 100 nmol/ml. Protein concentrations were measured by use of a protein assay kit from Bio-Rad Laboratories (Hercules, CA, USA) in accordance with the manufacturer’s instructions.
Analysis of Alveolar Bone
Skulls were separated and gingival tissue removed. Alveolar bones were defleshed for evaluation of bone loss [31]. Briefly, the mouse heads were removed, the alveolar bones were separated from the skull, and mandibles and maxillas were defleshed and stained. Digital photographs were prepared under stereomicroscopy equipped with a custom made stage holder and the images analyzed using NIH Image J software with inversion algorithm enhancements to capture the alveolar bone-loss area. The enclosed area highlighted the area of bone loss from CEJ to the horizontal bone level and the pixels were converted to mm2. Mean areas of bone loss in maxillas and mandibles were determined for each animal for comparisons between treatment and control groups.
Statistical Analysis
All results are expressed as mean ± SEM unless otherwise stated. Data were evaluated by analysis of variance, followed by appropriate post hoc test using GraphPad Instat version 3 for Windows (GraphPad Software, San Diego, CA, USA). Statistical significance was set at P < 0.05.
Results
DSS and TNBS mice tolerated oral application of these compounds for the duration of study with no mortality or substantial side effects estimated via general appearance. All experimental animals displayed body weight gain compared with their initial weight (Fig. 1).
DSS-treated animals developed clinical features of disease including diarrhea and bloody stools, with pale mucosa and anemia and did not gain weight during DSS administration but returned to normal during the weeks of abstinence. Animals treated with TNBS orally did not develop any systemic clinical symptoms and gained weight similar to sham controls, indicative of more local inflammatory response. Chronic colitis was manifest with increased splenic length (P < 0.05; Fig. 2) and weight (Fig. 3) in DSS-treated animals in a time-dependent fashion (weeks 7 and 12 P < 0.05; week 18 P < 0.01), because of general immune and inflammatory responses; cellular infiltration, however, remained normal in TNBS-treated mice (vs control P > 0.05 NS). Colonic tissue from DSS-treated mice became friable and significantly shortened (P < 0.001; Fig. 4) and colonic weight (Fig. 5) and pathological scores (3.5 ± 0.5 P < 0.001) increased because of increases in inflammatory cell populations and collagen deposits compared with normal controls (0) or TNBS-treated animals (0). In addition to colonic shrinkage, DSS-treated mice developed extensive dilation of the stomach wall, ileum, and megacolon, and displayed extensive abdominal fat deposits. The DSS-treated animals also showed dysregulated hepatic concentrations of endogenous antioxidants (cysteine, cystine, GSH, SAMe), measured by HPLC, demonstrating a profound systemic response (Figs. 6, 7, 8, 9). The concentration of liver cysteine (GSH precursor) increased (Fig. 6) whereas the liver cystine level decreased by a factor of two in DSS-treated animals compared with TNBS-treated or sham controls (weeks 12 and 18 P < 0.001; Fig. 7). However, liver GSH, the main source of intercellular antioxidant, was reduced in TNBS-treated animals (weeks 7, 12, and 18 P < 0.05) compared with other groups (Fig. 8). Unexpectedly, significant increases in levels of liver SAMe, a GSH precursor and methylating agent (weeks 12 and 18, Fig. 9), and the ratio of reduced (GSH) to oxidized (GSSG) glutathione (control 8.4 ± 1.2; TNBS 7.4 ± 4.6; DSS 9.2 ± 0.8) were observed in DSS-treated animals compared with normal or TNBS-treated animals. In addition, liver adenosine concentrations (Fig. 10) were reduced in treated animals compared with normal controls (TNBS P < 0.05, DSS P < 0.01).

Animals’ body weight gain during the 18 weeks; it was less prominent in DSS-treated groups but did not reach significance

Splenic length increased significantly in animals treated with DSS (7, 12, and 18 weeks P < 0.05) compared with TNBS-treated animals or controls (n = 5 animals/treatment/time point)

Splenic weight increased significantly in animals treated with DSS (7 and 12 weeks P < 0.05, 18 weeks P < 0.01) compared with TNBS-treated animals or controls (n = 5 animals/treatment/time point)

Colonic length measured at 7 weeks, 12 weeks, and 18 weeks was significantly reduced in DSS-treated mice (P < 0.001) compared with other groups (n = 5/time point/group; mean ± SEM)

Colonic weight measured at 7 weeks, 12 weeks, and 18 weeks was significantly increased in DSS-treated mice (P < 0.05) compared with other groups (n = 5/time point/group; mean ± SEM)

Liver cysteine concentration was increased significantly by DSS treatment (n = 5/time point/group; mean ± SEM)

Liver cystine concentration was reduced significantly by DSS treatment (P < 0.001 weeks 12 and 18; n = 5/time point/group; mean ± SEM)

Liver glutathione concentration was reduced significantly by TNBS treatment (n = 5/time point/group; mean ± SEM)

Liver SAMe concentration was increased significantly in DSS-treated animals (weeks 12 and 18)

Liver adenosine concentration was reduced by DSS treatment (weeks 12 and 18)
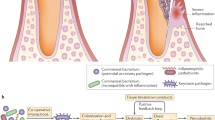
Administration of these inflammatory chemicals resulted in oral inflammation with significant increases in alveolar bone resorption in a time-dependent manner. The bone loss was detected as early as week 7, consistent with the development of chronic colitis in the DSS administered mice. The alveolar bone resorption progressed significantly to more severe periodontal bone loss (171 ± 5% of controls in DSS; 183 ± 8% of controls in TNBS animals; P < 0.001) through week 18.
Finally, mice were injected via gingival tissue (sub-mucosal) into the right maxilla with TNBS and sham challenged into the left maxilla using an inverted hypodermic needle. Alveolar bone resorption assessment was measured morphometrically 48–72 h after the injections. TNBS sub-mucosal delivery caused severe localized inflammation and granuloma formation. A significant increase in inflammatory cells infiltration (granuloma) and a 60% increase in bone resorption (P < 0.001) was observed in the TNBS-treated maxilla (0.980 ± 0.037 vs. 11.562 ± 0.165 mm2). In contrast untreated maxillas and mandibles showed no significant bone loss in the same animals. TNBS sub-mucosal localized delivery did not cause any colitic symptoms in these animals.
Discussion
This study demonstrates the induction of oral inflammation in mice periodically administered DSS or TNBS orally for a period of 18 weeks. Administration of the low dose DSS caused alveolar bone loss and chronic colitis as evidenced by severe shrinkage of the colonic tissue, infiltration of inflammatory cells into the colonic tissue, and periodontal alveolar bone involvement. The oral TNBS treatment exerted only a local effect on periodontal tissues leading to alveolar bone loss in a time-dependent manner with no intestinal manifestations.
IBD is regarded as an autoimmune response to commensal gut microbiota, and toxic agents including LPS, bypassing the compromised epithelium by underlying dysregulated oral tolerance [32]. The chronic inflammation occurring in IBD resembles the chronic immunoinflammatory response in the oral cavity that destroys the periodontium, leading to periodontitis and potential exfoliation of the teeth [20]. Although the occurrence of oral complications in IBD, especially in Crohn’s patients, is well-documented, few studies are available regarding the destruction of the periodontium in these individuals [33]. Existing studies have provided mixed findings regarding the expression of periodontal lesions in IBD patients. One study suggested approximate 12% higher prevalence of periodontal lesions in IBD compared with data from assessment of the oral health of United States adults [34].. About 20–60% of patients suffering from gastrointestinal syndromes also had oral mucosal manifestation of labial fissures, glossitis, or aphthous stomatitis, in addition to gingivitis and periodontitis [21]. In another trial, IBD patients had higher prevalence of periodontitis with smoking as modifier. Among non-smokers, both Crohn’s and ulcerative colitis patients showed deeper pockets compared with controls [17]. However, a separate IBD trial found that while IBD patients exhibited a significantly higher number of oral disease symptoms, periodontal findings were comparable with those of control patients [20]. IBD has also been reported as contributing to persistent oral lesions in pediatric populations [18], and suggesting that young patients with gingival and/or other oral lesions (e.g. orofacial granulomatosis symptoms) with or without other clinical GI symptoms should be evaluated for Crohn’s disease [19, 35, 36] In fact, it has been proposed that dentists can play a critical role in the early diagnosis of IBD, in helping to prevent general disease complications, and in improving the prognosis [19]. Thus, these clinical reports indicate the significance of oral complications associated with IBD diagnosis in patients [20].
At the molecular level, there are similarities in biological mechanisms of chronic inflammation and loss of function of GI tissues and the periodontium. Oxidant-mediated injury plays an important role in the pathophysiology of chronic inflammatory diseases including periodontitis [4–7] and IBD [1–3]. Plasma and gingival crevicular fluid (GCF) from patients with periodontitis contained reduced GSH and total antioxidant capacity [6], and lower antioxidant capacity of saliva with increased protein oxidation [37], consistent with dysregulation in oxidants and antioxidants contributing to the disease process. These ROS mediators also enhance production of numerous pro-inflammatory biomolecules, including IL-1β, IL-6, TNFα, cyclooxygenase 2, and osteopontin, among a broad array of molecules that have been reported to be elevated in GCF and tissues, either as hallmark or etiologic of the process [38–40]. These mediators are also frequently reduced after periodontal therapy [41]. These inflammatory markers are increased in IBD patients [42–44] and in animal models of this disease(s) [2, 3, 24–26, 28, 43, 45]. Previous reports indicate that upregulation of NF-κB responsive genes (e.g. TNFα) negatively correlated with down regulation of antioxidant activities in these models, indicating the role of ROS activation in gut inflammation. Similarly, therapeutic modalities with antioxidants, (i.e. PTCA and SAMe) increased blood levels of rGSH (intrinsic antioxidants), attenuated TNFα levels, and reduced inflammation in colitis [25, 26] and hepatitis models [46–48].
Long term administration of DSS to mice caused dysregulation of liver antioxidants. As expected, cystine and adenosine levels were significantly reduced in DSS-treated animals, with no major difference between controls and the TNBS-treated group. While unexpected, the concentration of cysteine, the ratio of reduced (GSH) to oxidized (GSSG) glutathione, and the endogenous multifunctional antioxidant (GSH precursor) SAMe were increased in these long term DSS-treated animals. One explanation could be that these animals were euthanized at the end of a DSS abstinence period, and hepatic tissues could be compensating for the loss of antioxidants caused by the previous DSS assault. A recent published article also observed increases of SAMe in response to LPS-induced toxicity in mice, resulting from an increase in SAMe biosynthesis and/or blocking of transmethylation [48]. Our previous studies have consistently shown a significant decrease in blood levels of reduced GSH during active disease in DSS-colitic mice and systemic effects of DSS-acute colitis were manifest with increased blood inflammatory cytokines, acute phase protein serum amyloid A responses, and deceases in the concentration of blood and liver antioxidants (GSH, cysteine) [24–26].
Implementation of the DSS and TNBS models should, then, enable studies of osteoimmunological interactions in the oral cavity. Oral topical administration of TNBS resulted in localized oral inflammation and alveolar bone resorption. However, TNBS injection into gingival tissues caused localized but severe infiltration of inflammatory cells, granuloma formation, and rapid and extensive alveolar bone loss. Implementation of the TNBS model of chronic inflammatory bone resorption will enable comparison of the contribution of ROS to inflammatory disease lesions in the oral cavity.
The chronic inflammation and tissue destruction in the gingival tissues is similar to those reported for IBD, targeting the innate immune system with DSS and the T-cell mediated immune responses with TNBS. These findings suggest these chemicals cause similar alterations of host responses at oral mucosal surfaces and provide a useful model system for examining periodontal inflammation and bone loss. Implementation of the DSS and TNBS models should, then, enable studies of osteoimmunological interactions in the oral cavity.
We believe that long-term administration of DSS mimics some aspects of disease progression in chronic IBD patients who suffer over prolonged periods of their lives. We suggest that this long-term model of IBD has some value for the study of late-stage mechanisms of IBD pathogenesis for development of new advanced therapeutic modalities for this condition.
References
Oz HS, Ebersole JL. Application of prodrugs to inflammatory diseases of the gut. Molecules. 2008;13:452–474. (review).
Neuman MG. Immune dysfunction in inflammatory bowel disease. Transl Res. 2007;149:173–186. (review).
Byme FR, Viney JL. Mouse models of inflammatory bowel disease. Curr Opin Drug Discov Devel. 2006;9:207–217. (review).
Chapple IL. Reactive oxygen species and antioxidants in inflammatory diseases. J Clin Periodontol. 1997;24:287–296.
Kesavalu L, Bakthavatchalu V, Rahman MM, et al. Omega-3 fatty acid regulates inflammatory cytokine/mediator messenger RNA expression in Porphyromonas gingivalis-induced experimental periodontal disease. Oral Microbiol Immunol. 2007;22:232–239.
Chapple IL, Brock G, Eftimiadi C, Mathews JB. Glutathione in gingival crevicular fluid and its relation to local antioxidant capacity in periodontal health and disease. J Clin Pathol Mol Pathol. 2002;55:367–373.
Waddington RJ, Moseley R, Embery G. Reactive oxygen species: A potential role in the pathogenesis of periodontal diseases. Oral Dis. 2000;6:138–151.
Marangon K, Devaraj S, Tirosh O, Packer L, Jialal I. Comparison of the effect of alpha-lipoic acid and alpha-tocopherol supplementation on measures of oxidative stress. Free Radic Biol Med. 1999;27:1114–1121.
Kim HJ, Chang EJ, Kim HM, et al. Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumor necrosis factor-alpha. Free Radic Biol Med. 2006;40:1483–1493.
Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: The “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122.
Enwonwu CO. Cellular and molecular effects of malnutrition and their relevance to periodontal diseases. J Clin Periodontol. 1994;21:643–657.
Enwonwu CO. Interface of malnutrition and periodontal diseases. Am J Clin Nutr. 1995;61:430S–436S.
Kawashi Y, Jaccard F, Cimasoni G. Sulcular polymorphonuclear leukocytes and gingival exudates during experimental gingivitis in man. J Periodontol Res. 1980;15:151–158.
Gustafsson A, Asman B. Increased release of free oxygen radicals from peripheral neutrophils in adult periodontitis after Fc delta-receptor stimulation. J Clin Periodontol. 1996;23:38–44.
Key LL Jr, Wolf WC, Gundberg CM, Ries Wl. Superoxide and bone resorption. Bone. 1994;15:431–436.
Makins R, Ballinger A. Gastrointestinal side effects of drugs. Expert Opin Drug Saf. 2003;2:421–429. (review).
Brito F, de Barros FC, Zaltman C, et al. Prevalence of periodontitis and DMFT index in patients with Crohn’s disease and ulcerative colitis. J Clin Periodontol. 2008;35:555–560.
Galbraith SS, Drolet BA, Kugathasan S, Paller AS, Esterly NB. Asymptomatic inflammatory bowel disease presenting with mucocutaneous findings. Pediatrics. 2005;116:e439–e444.
Ojha J, Cohen DM, Islam NM, Stewart CM, Katz J, Bhattacharyya I. Gingival involvement in Crohn disease. J Am Dent Assoc. 2007;138:1574–1581.
Grössner-Schreiber B, Fetter T, Hedderich J, Kocher T, Schreiber S, Jepsen S. Prevalence of dental caries and periodontal disease in patients with inflammatory bowel disease: A case–control study. J Clin Periodontol. 2006;33:478–484.
Mdinaridze GN, Rumiantsev VG, Maksimovskiĭ IM, Iurkov MI. State of the mouth cavity in patients with inflammatory intestinal diseases]. Eksp Klin Gastroenterol. 2006;4:17–21, 114.
Rezaie A, Ghorbani F, Eshghtork A, et al. Alterations in salivary antioxidants, nitric oxide, and transforming growth factor-beta 1 in relation to disease activity in Crohn’s disease patients. Ann N Y Acad Sci. 2006;1091:110–122.
Heitz-Mayfield LJ, Schätzle M, Löe H, et al. Clinical course of chronic periodontitis. II. Incidence, characteristics and time of occurrence of the initial periodontal lesion. J Clin Periodontol. 2003;10:902–908.
Oz HS, Chen T, de Villiers W, McClain C. Metallothionein overexpression does not protect against inflammatory bowel disease in a DSS murine colitis model. Med Sci Monit. 2005;11:BR69–BR73.
Oz HS, Chen TS, McClain CJ, de Villiers WJ. Antioxidants a novel therapy in a murine model of colitis. J Nutri Biochem. 2005;16:297–304.
Oz HS, Chen T, Nagasawa H. Comparative efficacies of two cysteine prodrugs and a glutathione delivery agent in a colitis model. Transl Res. 2007;150:122–129.
Ardite E, Sans M, Panes J, Romero FJ, Pique JM, Fernandez-Checa JC. Replenishment of glutathione levels improves mucosal function in experimental acute colitis. Lab Invest. 2000;80:735–744.
Oz HS, Zhong J, de Villiers W. Osteopontin ablation protects against progression of acute and chronic stages of TNBS-induced colitis. Gastroenterol Supl. 2008;134:A-525, T1296.
Garcia-Lafhente A, Antolin M, Guarner F, et al. Incrimination of anaerobic bacteria in the induction of experimental colitis. Am J Physiol. 1997;272:G10–G15.
Axelsson LG, Landström E, Bylund-Fellenius AC. Experimental colitis induced by dextran sulfate sodium in mice: Beneficial effects of sulfasalazine and olsalazine. Aliment Pharmacol Ther. 1998;12:925–934.
Oz HS, Ebersole JL. A novel murine model for chronic inflammatory alveolar bone loss. J Periodont Res. (in press).
Brandtzaeg P. Inflammatory bowel disease: Clinics and pathology. Do inflammatory bowel disease and periodontal disease have similar immunopathogeneses? Acta Odontol Scand. 2001;59:235–243.
Colella G, Riegler G, Lanza A, Tartaro GP, Russo MI, Tartaglione M. Changes in the mouth mucosa in patients with chronic inflammatory intestinal diseases. Minerva Stomatol. 1999;48:367–371.
Flemmig TF, Shanahan F, Miyasaki KT. Prevalence and severity of periodontal disease in patients with inflammatory bowel disease. J Clin Periodontol. 1991;18:690–697.
Girlich C, Bogenrieder T, Palitzsch KD, Schölmerich J, Lock G. Orofacial granulomatosis as initial manifestation of Crohn’s disease: A report of two cases. Eur J Gastroenterol Hepatol. 2002;14:873–876.
Sigusch BW. Periodontitis as manifestation of Crohn’s disease in primary dentition: A case report. J Dent Child (Chic). 2004;71:193–196.
Sculley DV, Langley-Evans SC. Salivary antioxidants and periodontal disease. Proc Nutr Soc. 2002;61:137–143.
Bretz WA, Weyant RJ, Corby PM, et al. Systemic inflammatory markers, periodontal diseases, and periodontal infections in an elderly population. J Am Geriatr Soc. 2005;53:1532–1537.
Ebersole JL, Taubman MA. The protective nature of host responses in periodontal diseases. Periodontol 2000. 1994;5:112–141.
Garlet GP, Martins W Jr, Fonseca BA, Ferreira BR, Silva JS. Matrix metalloproteinases, their physiological inhibitors and osteoclast factors are differentially regulated by the cytokine profile in human periodontal disease. J Clin Periodontol. 2004;31:671–679.
Sharma CG, Pradeep AR. Gingival crevicular fluid osteopontin levels in periodontal health and disease. J Periodontol. 2006;77:1674–1680.
Mishima R, Takeshima E, Sawi K, et al. High plasma opsteopontin levels in patients with inflammatory bowel disease. J Clinic Gasteroentrol. 2007;41:162–172.
Zhong J, Eckhart E, Oz HS, Bruemmer D, de Villiers W. Osteopontin deficiency protects mice from DSS-induced colitis. Inflamm Bowel Dis. 2006;12:790–796.
Pizarro TT, Cominelli F. Cytokine therapy for Crohn’s disease: Advances in translational research. Annu Rev Med. 2007;58:433–444.
Schwartz L, Abolhassani M, Pooya M, et al. Hyperosmotic stress contributes to mouse colonic inflammation through the methylation of protein phosphatase 2A. Am J Physiol Gastrointest Liver Physiol. 2008;295:G934–G941.
Oz HS IMH, Chen T, de Villiers W, McClain C. Glutathione enhancing agents protect against Steatohepatitis in a model. J Biochem Mol Toxicol. 2006;20:39–47.
Oz HS, McClain CJ, Nagasawa HT, Ray MB, de Villiers WJ, Chen TS. Diverse antioxidants protect against acetaminophen hepatotoxicity. J Biochem Mol Toxicol. 2004;18(6):361–368.
Ko K, Yang H, Noureddin M, et al. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab Invest. 2008;88:1121–1129.
Acknowledgments
Marcia C. Liu provided HPLC technical assistance. This research was supported by National Institutes of Health grants NCCAM-AT1490 and NIDCR-DE19177 (HO).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oz, H.S., Chen, T. & Ebersole, J.L. A Model for Chronic Mucosal Inflammation in IBD and Periodontitis. Dig Dis Sci 55, 2194–2202 (2010). https://doi.org/10.1007/s10620-009-1031-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-009-1031-x