
Abstract
Amniotic fluid (AF) contains heterogeneous and multipotential cell types. A pure mesenchymal stem cells group can be sorted from AF using flow cytometry. In order to evaluate a possible therapeutic application of these cells, the human AF-derived c-kit+ stem cells (c-kit+ AFS) were compared with the c-kit− (unselected) stem cells (c-kit− AFS). Our findings revealed that the optimal period to obtain c-kit+ AFS cells was between 16 and 22 weeks of gestation. Following cell sorting, c-kit+ AFS cells shared similar morphological and proliferative characteristics as the c-kit− AFS cells. Both c-kit+ and c-kit− AFS cells had the characteristics of mesenchymal stem cells through surface marker identification by flow cytometric and immunocytochemical analysis. Both c-kit+ and c-kit− AFS cells could differentiate along adipogenic and osteogenic lineages. However, the myocardial differentiation capacity was enhanced in c-kit+ AFS cells by detecting GATA-4, cTnT, α-actin, Cx43 mRNA and protein expression after myocardial induction; whereas induced c-kit− AFS cells were only detected with GATA-4 mRNA and protein expression. The c-kit+ AFS cells could have potential clinical application for myogenesis in cardiac regenerative therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is widely agreed that regenerative capacity of human myocardium is grossly inadequate to compensate for the severe loss of heart muscle as seen in myocardial infarction and heart failure, which is a major cause of mortality and morbidity worldwide (Lloyd-Jones et al. 2010). Cellular cardiomyoplasty has the potential to be an ideal method for the treatment of heart failure compared with other current therapeutic modalities, including medical therapy, mechanical assist devices, and cardiac transplantation, all of which have a number of limitations. Within the last 20 years, stem cells based therapies, including adult stem cells and embryonic stem cells, have been employed towards this end due to their ability to self-renew and to differentiate into multiple cell types. Stem cells from many sources have been explored for their advantages and limitations in clinical use. Adult human bone marrow contains a rare population of mesenchymal stem cells (MSCs) (0.01–0.001%) (Castro-Malaspina et al. 1980; Civin et al. 1996). They can be extensively expanded in vitro and, then cultured under specific permissive conditions to differentiate into osteogenic, adipogenic, chondrogenic, muscle, neuronal and stromal cells (Pittenger et al. 1999; Deans and Moseley. 2000). However, adult stem cells have poorly characterized growth conditions and cannot be effectively propagated in vitro, limiting their usefulness in acute myocardial infarction (MI) treatment as the optimal transplantation time was shown to be 7–10 days after acute MI (Peruga et al. 2009; Tendera et al. 2009; Ripa et al. 2006). Therefore, allogeneic transplantation method needs to be developed to address the issue of narrow transplantation time window in acute MI. Amniotic fluid (AF) is an important alternative source of fetal stem cells. AF contains a heterogeneous population of cells, which are contributed mainly from the fetal skin, digestive, respiratory and urinary tract, and the placental membranes. De Coppi et al. (2007) used immunoselection with magnetic microspheres to isolate the CD117+ (c-kit) population (approximately 1% of total AF cells) from many amniocentesis specimens and found that these cells can be readily expanded in culture as stable lines. They have routinely established clonal AFS cell lines with a typical doubling time of about 36 h and no need for feeder layers. Over 90% of the cells expressed the transcription factor Oct4. The surface marker profile of the AF stem cells and their expression of the transcription factor Oct4 suggests that they represent an intermediate stage between pluripotent embryonic stem (ES) cells and lineage-restricted adult stem cells (Martin 1981; Evans and Kaufman. 1981). The differentiation potential of these cells is also considered to exceed all the multipotent adult stem cells (De Coppi et al. 2007). Unlike human ES cells, AFS cells do not form tumors in vivo. A low risk of tumorigenicity would be advantageous for eventual therapeutic applications. Through c-kit, the surface marker. A high purity AF cell population can be sorted using c-kit as a target, and it is considered that a purer cell population is safer for clinical use (Phermthai et al. 2010). However, c-kit-based selection procedure may induce a different mesenchymal differentiation capacity from the unselected cell populations (Arnhold et al. 2011). Stefano (Da Sacco et al. 2010) noted that the composition of AF cells has a precise timetable during gestation. Therefore, in our study we have investigated the growth and phenotypic characteristics of AF cells in a long range of gestation time by describing growth curves and identifying the cells surface markers. The focus of our study was the differentiation potential of AF cells after a cell sorting procedure using the cell surface marker c-kit. Our data revealed that AF cells from the c-kit positive samples showed improved myocardial differentiation characteristics, while the osteogenic and adipogenic differentiation capacities were similar in c-kit+ and c-kit− AFS cells through detect the expression of related genes.
Materials and methods
Samples and origin of human amniotic fluid cells
We obtained 13 human amniotic fluid samples by amniocentesis performed between 16 and 31 weeks of gestation. Eight samples were from donors who needed prenatal diagnosis and other five samples were obtained prior to intrauterine ethacridine lactate administration for individuals undergoing legal termination. This is an ethically approved and accepted procedure in the first affiliated hospital of Chinese PLA general hospital. All patients have signed informed consents. The average age of the donors were 27.36 ± 7.42 years with an average body weight of 68.73 ± 18.64 kg. Cell samples were only used when no major abnormalities were revealed by the cytogenetic analysis.
Isolation, culture and sorting of amniotic fluid cells
Cells were isolated from the AF samples no more than 4 h prior to being used. The samples were centrifuged at 1,500 rpm for 5 min. After removal of the supematant, the cells were cultured at 37 °C in a humidified incubator containing 5% CO2. The AF cells culture medium was composed of minimum essential medium (α-MEM; Gibco, Langley, OK, USA) supplemented with 20% Chang Medium (18% Chang B plus 2% Chang C; Irvine Scientific, Santa Ana, CA, USA), 20% embryonic Stem fetal bovine serum (ES FBS; Gibco, Langley, OK, USA), 1% glutamine and 1% penicillin/streptomycin. After 3 days, non-adherent cells and debris were discarded and the adherent cells were continued to be cultivated in pre-confluency. The morphologic characteristics of the AF cells were observed with inverted phase contrast microscope (Olympus, Tokyo, Japan). When the cells reached 80–90% confluency, cells were dissociated by trypsin–EDTA solution (Invitrogen, San Francisco, CA, USA) and then sorted by flow cytometry for c-kit+ cells using a monoclonal anti-c-kit (CD117) antibody (Becton Dikinson Berkeley, CA, USA). The c-kit+ cells were expanded and subsequently cloned by limiting dilution and cultured in sub-confluent conditions in the presence of AF cells medium.
Growth curves
The c-kit+ and c-kit− AF cells were trypsinized and inoculated in 96-well culture plates to a total volume of 100 μl/well (3,000 cells/well) and incubated at 37 °C in a humidified incubator containing 5% CO2. These cells were allowed to attach overnight and then incubated for seven days. Control wells for absorbance readings contained no cells except for the culture medium. Cell viability was determined by MTT cytotoxic assay. Triplicate wells were supplemented with 20 μl MTT solution every day. After 4 h incubation the absorbance was measured at 490 nm using an enzyme micro-plate reader (Bio-Rad Laboratories, Berkeley, CA, USA).
Flow cytometry
The specific cell surface antigens of the c-kit+ and c-kit− AF cells at passage 5–7 were characterized by flow cytometric analysis. Cells in culture were trypsinized, washed and resuspended in phosphate-buffered saline (PBS; Sigma-Aldrich Louis, MO, USA) at concentration of 1 × 105 cells/100 μl. Cells were stained directly with FITC-immunolabeled mouse anti-human monoclonal antibodies against CD29, CD44, CD73, CD90, CD105, CD34, CD45, HLA-ABC, HLA-DR (Becton Dikinson, Berkeley, CA, USA) and rabbit anti-human antibodies against Oct4, Sox2 and Nanog (Becton Dikinson). Analysis was performed with flow cytometer (Cytomics FC 500 MPL; Bechman Coulter, Brea, CA, USA) and data were analyzed with the Cxp Software. Data were expressed as number of cells/106 cytometric events.
Immunocytochemical analysis
To monitor cytoplasmic antigens, c-kit+ and c-kit− AF cells were collected separately using a centrifuge (Kubota Corporation, Naniwa-ku, Osaka, Japan). The primary monoclonal antibodies used in this study were anti-CD29, anti-CD34, anti-CD44, anti-CD45, anti-CD73, anti-CD90, anti-CD105, anti-HLA-ABC, anti-HLA-DR, anti-Oct-4, anti-Sox2, and anti-Nanog. Cells were washed, fixed, and incubated with 1:100 mouse anti-human primary monoclonal antibodies overnight at 4 °C. Secondary antibodies FITC-conjugated horse anti-mouse IgG (Invitrogen, San Francisco, CA, USA) were added at room temperature for 1 h. Cell nuclei were counterstained with 1 μg/ml 4′,6-diamino -2-phenylindole (Vectashild) in PBS for 5 min and mounted in mounting medium. Negative controls were performed by omitting the primary antibodies. All images were captured using a confocal laser scanning microscope (Olympus Fluoview fv1000).
Multipotent differentiation
Cells were analyzed for their mesenchymal differentiation capacity towards the osteogenic and adipogenic lineages. Adipogenic differentiation was induced by culturing AFS cells for 3 weeks in DMEM low-glucose medium containing 10% FBS, 10−6 M dexamethasone, 0.5 mM isobutyl-methyl xanthine (IBMX), 200 μM indomethacin, and 10 μg/ml insulin. Adipogenic differentiation was evaluated for the accumulation of lipid vacuoles via staining with Oil-Red O. Reverse transcription polymerase chain reaction (RT-PCR) was also performed to detect the expression of adiponectin gene. Osteogenic differentiation was induced by culturing c-kit+ and c-kilt− AFS cells for 3 weeks in osteogenic medium (OM). OM consists of DMEM low-glucose medium containing 10% FBS, 10−8 M dexamethasone, 10 mM β-glycerophosphate and 0.2 mM ascorbic acid. To assess calcium accumulation, cells were stained with Von Kossa stain, TR-PCR was performed to detect osteopontin mRNA. The primers used in this study were based on published human sequences (Table 1).
Myocardium-like cells differentiation
The c-kit+ and c-kit− AFS cells were respectively cocultured with rat neonatal cardiomyocytes at a ratio of 1:10 in a transwell system, which allowed the diffusion of secreted factors but prevented cell contact between the two cell types (Heng et al. 2004). The cells mixture was cultured at 1 × 105 cells/well in Dulbecco’s modified Eagle’s medium (Gibco, Langley, Oklahoma, USA) supplemented with 5% chick embryo extract (Sigma), 10% horse serum (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin. After 10 days of coculture with neonatal cardiomyocytes, RNA was extracted from AFS cells for real-time PCR analysis. Undifferentiated AFS cells were used as controls.
RT-PCR and real-time PCR
The total mRNA was extracted with Trizol Reagent (Invitrogen, San Francisco, CA, USA) from the c-kit+ and the c-kit− cultured cells. PCR reactions were carried out by mixing 1 μl of cDNA template, 250 nM of each primer, 200 μM dNTP mixture and 1 U of Taq DNA polymerase in a volume of 20 μl. Samples were amplified in a thermocycler. Following PCR, 10 μl of the PCR product was electrophoresed on a 1% agarose gel and imaged using a gel imaging instrument. For real-time PCR, transcript levels were standardized to the corresponding human GAPDH level. Amplification data were collected using the ABI PRISM 7,900 and analyzed using the Sequence Detection System 2.0 software. Primer information is provided in Supplementary Material Table 1.
Western blot analysis
For western blot analyses, protein extracts were prepared in buffer containing 20 mmol/l HEPES pH 7.9, 0.4 mol/l NaCl, 2.5% glycerol, 1 mmol/l EDTA, 1 mmol/l phenylmethylsulfonyl fluoride, 0.5 mmol/l NaF, 0.5 mmol/l Na3VO4, 0.02 μg/ml leupeptin, 0.02 μg/ml aprotinin, 0.003 μg/ml benzamidine chloride, 0.1 μg/ml trypsin inhibitor and 0.5 mmol/l dithiothreitol (DTT). Proteins were resolved on denaturing SDS-polyacrylamide gels (10%), transferred to Immuno-Blot poly-vinylidene difluoride membranes (Bio-Rad Laboratories, Berkeley, CA, USA). Western blots were probed with goat anti-human primary antibodies (anti-GATA-4, anti-cTnT, anti-α-actin, and anti-Cx43). Primary antibodies binding was detected with horseradish peroxidase-conjugated rabbit anti-goat (Zymed Laboratories, South San Francisco, CA, USA) and visualized using the ECL plus Western Blotting Detection System (Bio-Rad Laboratories).
Statistical analysis
Statistical analysis was performed with the SPSS10.0 statistics software (SPSS Inc., Chicago, IL, USA). Data are presented as mean ± SD. Data were analyzed for statistical significance by Student’s t test. P value <0.05 was considered significant.
Results
Morphology and proliferation characteristics
The total of 13 AF samples (A–M) of 16–31 weeks of gestation were analyzed (Table 2). Freshly cultured primary AF cells were morphologically heterogeneous (Fig. 1a–c). After cultured for several days, an adherent cell population could be seen. The average time to reach adherence was 5.12 ± 1.87 days. After the first passage, a homogenous cell layer (monolayer) could be seen, however the cell population was rather heterogeneous. The adherent cells grew in islands or in cell groups showing elongated spindle shape or epithelial-like appearance (Fig. 1d–f). The c-kit+ AFS cells can only be sorted from AF cell adherent cultures showing fibroblast-like cells (Table 2). Following cell sorting, c-kit+ AFS cells grew conglomerately with a homogeneous fibroblastoid shape (Fig. 1j). Five AF samples of 16–22 weeks of gestation were positive for c-kit+ after sorting (named as c-kit+ AFS cells, Table 2). There were other 5 AF samples presented with fibroblast-like shape but did not yield c-kit+ AFS cells after c-kit sorting (named as c-kit− AFS cells, Table 2). The c-kit+ cells constituted 3.30 ± 1.24% of the adherent AF cells. Fibroblastoid cells were recultured and could proliferate for more than 50 passages in vitro. Three AF samples of later gestation at weeks 26, 30 and 31 (Table 2, sample J, L and M) were epithelioid-like and could not be cultured for more than five passages. These samples were not a good source of mesenchymal stem cells (Table 2). The proliferative characteristics were evaluated by MTT proliferation analysis. The cells were expanded for 7 days. There were no significant differences between c-kit+ and c-kit− AFS cells from passage 5 and passage 10 for they had the similar growth curves (Fig. 2).

Morphological characteristics of AF cells. 1, 2 and 3 were three representative samples of human AF cells. Freshly cultured AF cells were a heterogeneous population in suspension (a–c, ×200 magnification). Cultured AF cells began to adhere after being cultured for 3–7 days and showed elongated spindle shape or epithelial-like appearance (d–f, ×200 magnification). After continuous culture, the morphology of the cells presented significant difference: some were mainly fibroblast-like (g–h, ×200 magnification, black arrows showing fibroblast-like cells) and others were mainly epithelial-like (i, ×200 magnification, red arrows showing epithelial-like cells). Following the c-kit sorting procedure, sample 1 was successfully sorted of c-kit+ AFS cells, however, sample 2 and 3 did not yield significant c-kit+ AFS cells. The sorted c-kit+ AF cells were recultured and grew conglomerately with a homogeneous fibroblastoid shape (j, ×200 magnification). (Color figure online)

Growth curves by MTT proliferation analysis. The growth curves of c-kit+ and c-kit− AFS cells in passage 5 and passage 10 were similar. Plateau phase was not reached in these 7-day cultures
AF cells gene expression characterization
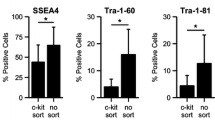
To better characterize AFS cells, we compared expression levels of several cell surface marker genes between the c-kit+ and c-kit− AFS cells at passage 5–7. Data from flow cytometry and immunocytochemical analysis revealed strong expression of CD29, CD44, CD45, CD73, CD90, CD105 and HLA-ABC in both two cell types. Trace levels of CD34, CD45 and HLA-DR were detected, being similar in both c-kit+ and c-kit− AFS cells (Table 2; Fig. 3). However, there were significant differences in the expression levels of the pluripotency markers using antibodies against Oct4, Sox2 and Nanog. The c-kit+ AFS cells showed high levels of expression in Oct4 (88.44%), Sox2 (91.1%) and Nanog (72.5%), while the c-kit− AFS cells were mostly negative in the expression of Oct4 (3.07%), Sox2 (0.55%) and Nanog (0.84%) (Fig. 4a). To further characterize the c-kit+ AFS cells, we compared the expression levels of Oct4, Sox2 and Nanog between c-kit+ and c-kit− AFS cells by RT-PCR and immunocytochemical analysis (Fig. 4b–c). The RT-PCR and immuno-cytochemical analysis confirmed the flow cytometry results that the c-kit+ AFS cells showed strong Oct4, Sox2 and Nanog expression, and the c-kit− AFS cells did not express these genes (Fig. 4).

Immunocytochemical analysis. Immunostaining was performed on c-kit+ and c-kit− AFS cells using antibodies against CD29, CD34, CD44, CD45, CD73, CD90, CD105, HLA-ABC and HLA-DR. Nuclei were stained with DAPI in all cells. All scale bars represent 100 μm

Pluripotency markers Oct4, Sox2 and Nanog expression in c-kit+ and c-kit− AFS cells. a Flow cytometry analysis. b RT-PCR analysis. Line 1 c-kit+ AFS cells; Line 2 c-kit− AFS cells. Data shown are representative of three independent experiments. c Immunocytochemical analysis. 1 Nuclear stain with DAPI (blue); 2 Antibody staining of Oct4, Sox2 and Nanog protein levels (green); 3 Fusion of 1 and 2. All scale bars represent 100 μm. (Color figure online)
Adipogenic and osteogenic differentiation
Following in vitro adipogenic induction, confocal laser scanning microscope showed accumulation of lipid vacuoles in both c-kit+ and c-kit− AFS cells and the cells were positive for Oil-Red O staining. Adiponectin gene expression was detected by RT-PCR in both cell types (Fig. 5). Following in vitro osteogenic induction, both c-kit+ and c-kit− AFS cells were shown to be positive for Von Kossa stain. Osteopontin gene expression was positive by RT-PCR in both cell types (Fig. 5).

Adipogenic and osteogenic differentiation of c-kit+ and c-kit− AFS cells. a Oil-Red O staining for lipid vacuoles accumulation after adipogenic differentiation in c-kit+ AFS cells (a) and c-kit− AFS cells (b). Von Kossa staining for mineralization after osteogenic differentiation in c-kit+ AFS cells (c) and c-kit− AFS cells (d), b RT-PCR for adiponectin and osteopontin expression after adipogenic and osteogenic differentiation. Adiponectin (187 bp): 1 c-kit+; 2 c-kit−; osteopontin (173 bp): 3 c-kit+; 4 c-kit−. (Color figure online)
Myocardium-like cells differentiation
Following in vitro myocardial induction, real-time PCR and Western blot showed that myocardial genes GATA-4, cTnT, α-actin, and Cx43 were detected in induced c-kit+ AFS differentiated cells, while the induced c-kit− AFS cells only showed mild GATA-4 expression and negative expression of cTnT, α-actin, and Cx43 (Fig. 6).

Myocardial induction in c-kit+ and c-kit− AFS cells. a Real-time PCR analysis of GATA-4, cTnT, α-actin and Cx43 mRNA expression were performed in c-kit + and c-kit- AFS cells. Results were shown as means ± SD from three independent experiments. 1 c-kit+ AFS cells before induction; 2 induced c-kit+ AFS cells; 3 c-kit− AFS cells before induction; 4 induced c-kit− AFS cells. b Western blot analysis of GATA-4, cTnT, α-actin and Cx43 protein expression were performed for c-kit+ and c-kit− AFS cells
Discussion
China has a high birth rate of more than 16 million neonates every year. Amniocentesis is performed as an routine clinical diagnosis technique for fetal genetic determination in prenatal diagnosis. Therefore, amniotic fluid (AF) can be readily harvested as a source of stem cells for potential clinical application. We obtained 13 AF samples of 16–31 weeks of gestation. The average vital cells number was 100–2,500 cells/ml (data not shown). We found that both epithelial-like and fibroblast-like cells appeared in the primary human AF culture. The average time to reach adherence was 5.12 ± 1.87 days. After 3–4 passages, most cells exhibited fibroblast-like appearance. These cells could proliferate for more than 50 passages in our culture study. There were three samples from the 26th, 30th and 32th week of gestation, which mainly grew epithelial-like cells instead of fibroblast-like cells and could not be propagated for more than five passages. This demonstrated that these cells were not MSCs for the International Society for Cellular Therapy position statement has given a definition of MSCs: (1) plastic adherence and fibroblast-like morphology; (2) multipotential and multilineage differentiation capacity; (3) expression of a typical set of surface markers such as CD73, CD90, and CD105; and (4) lack of lineage-specific markers such as CD34, CD14, CD45 (Dominici et al. 2006). Since these three samples were from the late stage of second-trimester, it suggested that AFS cell number may decrease as pregnancy progresses. In our experience, 16–24 weeks of gestation were an optimal period to culture AF cells to obtain stem cells, which showed a MSCs phenotype. The cultured AFS cells (both c-kit+ and c-kit− cells) were strongly positive for MSCs markers (CD29, CD44, CD73, CD90, CD105). The cultured c-kit+ and c-kit− AFS cells were negative for hematopoietic markers CD34 and CD45. Currently, there are different and even conflicting results of identifying stem cell surface markers obtained in different papers. These differences may be due to different proliferative stages of the cells in culture (Gronthos et al. 2001) and the donor’s heterogeneity. Mafi et al. (2011) have reviewed 29 studies looked at mesenchymal cells from different tissues and also identified various cell surface markers characterizing the cells. They found that CD105, CD90, CD44, CD73, CD29, CD13, CD34, CD146, CD106, CD45 and CD166 ranked among the most commonly reported positive cell surface markers on mesenchymal cells, and CD34, CD14, CD45, CD11b, CD49d, CD106, CD10 and CD31 were the most frequently absent surface markers in the mesenchymal stem cells. Our results were similar to these studies. In our study, Class I MHC antigens (HLA-ABC) were positive in both c-kit+ and c-kit− AFS cells. MHC Class II antigens (HLA-DR) were negative in both types of AFS cells. Both the c-kit+ and c-kit− AFS cells were shown to be non-immunogenic, due to lack of MHC class II antigen expression (Le Blanc and Ringdén 2005). This is undoubtedly a valuable characteristic of AFS cells which could reduce the need for immunosuppression following allogeneic transplantation.
Some researchers named the fibroblast-like cells as AF-derived MSCs. However, a homogenous cell population is needed for clinical use. In order to obtain a more homogenous cell population and to isolate mesenchymal stem cells, one option is to conduct cell sorting based on cell surface markers. C-kit plays an important role in gametogenesis, melanogenesis and hematopoiesis. This receptor protein is usually present on human ES cells and primordial germ cells (Da Sacco et al. 2010; Fleischman 1993). C-kit+ cells represent a group of purified MSCs newly found in AF. It is still not clear what is the origin of these cells and what their role is in fetal development. Several studies have revealed potentials in clinical use for these cells (Da Sacco et al. 2010; Fleischman 1993; Perin et al. 2010). De Coppi et al. (2007) showed that c-kit+ AFS cells were broadly multipotent, which could give rise to adipogenic, osteogenic, myogenic endothelial, neurogenic and hepatic lineages inclusive of all embryonic germ layers. However, the c-kit+ cells were reported to constitute only 1–5% of the total AF cells (De Coppi et al. 2007; Arnhold et al. 2011). We obtained a similar result of 3.30 ± 1.24% by flow cytometric sorting. Da Sacco et al. reported that c-kit+ AFS cells increased between 17 and 18 weeks of gestation, and disappeared at 19–20 weeks of gestation (Da Sacco et al. 2010). Our findings showed that c-kit+ AFS cells were present from 16–22 weeks of gestation. The discrepancy may partially be due to the heterogeneity of the original cell population. The c-kit+ and c-kit− cells showed similar characteristics in passage 5 and passage 10 as shown by the MTT assay. It suggested that both the c-kit+ and c-kit− AFS cells had good proliferate capacities even at the 10th passage.
In our study, Oct4, Sox2 and Nanog were more frequently positive in c-kit+ AFS cells than in c-kit− AFS cells. By flow cytometric analyses, Oct-4 was positive in 88.44% of c-kit+ AFS cells and in 3.07% of c-kit− AFS cells. Sox2 was positive in 91.1% of c-kit+ AFS cells and in 0.55% of c-kit− AFS cells. Nanog was positive in 72.5% of c-kit+ AFS cells and in 0.84% of c-kit− AFS cells. Oct4, Sox2 and Nanog have been associated with the maintenance of the undifferentiated state and the pluripotency of ES cells (Prusa et al. 2003). Our results suggest that the c-kit+ AFS cells possess more ES cell characteristics than c-kit− AFS cells even though both expressed MSCs surface makers. It suggests that there may be different differentiation capacities between the two types of cells. The mesenchymal differentiation capacity was evaluated by adipogenic and osteogenic induction in vitro for the c-kit+ and c-kit− AFS cells. Both types of cells were successfully differentiated into the osteogenic and adipogenic lineages. The osteogenic gene osteopontin and adipogenic gene adiponectin were detected by RT-PCR respectively.
Our data also showed that c-kit+ AFS cells have stronger myocardial-like cell differentiation capacity than c-kit− AFS cells by real-time PCR and western blot analysis. Two types of cells were cocultured with rat neonatal cardiomyocytes for cardiomyocyte induction (Yeh et al. 2010). The induced c-kit+ AFS cells were positive for GATA-4, cTnT, α-actin and Cx43 mRNA by real-time PCR analysis and corresponding protein expression by western blot analysis. However, the induced c-kit− AFS cells were only mildly positive for GATA-4 mRNA and protein expression. GATA-4 is an important transcription factor in early myocardial development. cTnT is a protein whose only known function is as part of the troponin complex of myofibrils in cardiac and embryonic skeletal muscles (Antin et al. 2002). Alpha-actin is one of the isoforms of the actin family found in muscle tissue (Oma and Harata 2011). Gap junctions play significant regulatory roles in embryonic development, electrical coupling, metabolic transport, apoptosis, differentiation, tissue homeostasis and carcinogenesis (Laird 2010). Cx43 is one of the gap junction proteins and is essential for the development of normal cardiac architecture and ventricular conduction. Therefore, our data revealed that the induced c-kit+ AFS cells had more myocardial cells characteristics including the stronger expression of GATA-4 and several mature myocardial genes c-TnT, α-actin and Cx43. In contrast, the induced c-kit− AFS cells expressed weaker GATA-4 levels than c-kit+ AFS cells and had no expression of the mature myocardial genes.
Conclusion
Our study demonstrates that AF cells from 16–22 weeks of gestation could be used for obtaining c-kit+ cell. We obtained pure stem cells population using c-kit surface markers by flow cytometric sorting. The c-kit+ and c-kit− AFS cells had similar morphology and proliferation characteristics, as well as mesenchymal differentiation capacity along the osteogenic and adipogenic lineages. The c-kit+ AFS cells had stronger myocardial-like cells differentiation capacity than c-kit− AFS cells. The c-kit+ AFS cells may possess more ES cell characterictics than c-kit− AFS with higher Oct-4, Sox2, and Nanog expression. The c-kit+ AFS cells have the potential in clinical application for myogenesis in cardiac regenerative therapy.
References
Antin PB, Bales MA, Zhang W, Garriock RJ, Yatskievych TA, Bates MA (2002) Precocious expression of cardiac troponin T in early chick embryos is independent of bone morphogenetic protein signaling. Dev Dyn 225:135–141
Arnhold S, Glüer S, Hartmann K, Raabe O, Addicks K, Wenisch S, Hoopmann M (2011) Amniotic-fluid stem cells: growth dynamics and differentiation potential after a CD-117-based selection procedure. Stem Cells Int 2011:715341
Castro-Malaspina H, Gay RE, Resnick G, Kapoor N, Meyers P, Chiarieri D, McKenzie S, Broxmeyer HE, Moore MA (1980) Characterisation of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood 56:289–301
Civin CI, Trischmann T, Kadan NS, Davis J, Noga S, Cohen K, Duffy B, Groenewegen I, Wiley J, Law P, Hardwick A, Oldham F, Gee A (1996) Highly purified CD34-positive cells reconstitute hematopoiesis. J Clin Oncol 14:2224–2233
Da Sacco S, Sedrakyan S, Boldrin F, Giuliani S, Parnigotto P, Habibian R, Warburton D, De Filippo RE, Perin L (2010) Human amniotic fluid as a potential new source of organ specific precursor cells for future regenerative medicine applications. J Urol 183:1193–1200
De Coppi P, Bartsch G Jr, Siddiqui MM, Xu T, Santos CC, Perin L, Mostoslavsky G, Serre AC, Snyder EY, Yoo JJ, Furth ME, Soker S, Atala A (2007) Isolation of amniotic stem cell lines with potential for therapy. Nat Biotechnol 25:100–106
Deans RJ, Moseley AB (2000) Mesenchymal stem cells: biology and potential clinical uses. Exp Hematol 28:875–884
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj, Horwitz E (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317
Evans MJ, Kaufman MH (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 292:154–156
Fleischman RA (1993) From white spots to stem cells: the role of the Kit receptor in mammalian development. Trends Genet 9:285–290
Gronthos S, Franklin DM, Leddy HA, Robey PG, Storms RW, Gimble JM (2001) Surface protein characterization of human adipose tissue-derived stromal cells. J Cell Physiol 189:54–63
Heng BC, Haider HKh, Sim EK, Cao T, Ng SC (2004) Strategies for directing the differentiation of stem cells into the cardiomyogenic lineage in vitro. Cardiovasc Res 62:34–42
Laird DW (2010) The gap junction proteome and its relationship to disease. Trends Cell Biol 20:92–101
Le Blanc K, Ringdén O (2005) Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 11:321–334
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J, American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2010) Executive summary: heart disease and stroke statistics-1010 update: a report from the American heart association. Circulation 121:948–954
Mafi R, Hindocha S, Mafi P, Griffin M, Khan WS (2011) Adult mesenchymal stem cells and cell surface characterization: a systematic review of the literature. Open Orthop J 2:253–260
Martin GR (1981) Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA 78:7634–7638
Oma Y, Harata M (2011) Actin-related proteins localized in the nucleus from discovery to novel roles in nuclear organization. Nucleus 2:38–46
Perin L, Sedrakyan S, Giuliani S, Da Sacco S, Carraro G, Shiri L, Lemley KV, Rosol M, Wu S, Atala A, Warburton D, De Filippo RE (2010) Protective effect of human amniotic fluid stem cells in an immunodeficient mouse model of acute tubular necrosis. PLoS ONE 5:e9357
Peruga J, Plewka M, Kasprzak J (2009) Intracoronary administration of stem cells in patients with acute myocardial infarction-angiographic follow-up. Kardiol Pol 67:477–484
Phermthai T, Odglun Y, Julavijitphong S, Titapant V, Chuenwattana P, Vantanasiri C, Pattanapanyasat K (2010) A novel method to derive amniotic fluid stem cells for therapeutic purposes. BMC Cell Biol 11:79
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Prusa AR, Marton E, Rosner M, Bernaschek G, Hengstschläger M (2003) Oct-4-expressing cells in human amniotic fluid: a new source for stem cell research? Hum Reprod 18:1489–1493
Ripa RS, Jørgensen E, Wang Y, Thune JJ, Nilsson JC, Søndergaard L, Johnsen HE, Køber L, Grande P, Kastrup J (2006) Stem cell mobilization induced by subcutaneous granulocyte-colony stimulating factor to improve cardiac regeneration after acute ST-elevation myocardial infarction result of the double-blind, randomized, placebo-controlled stem cells in myocardial infarction (STEMMI) trial. Circulation 113:1983–1992
Tendera M, Wojakowski W, Ruzyłło W, Chojnowska L, Kepka C, Tracz W, Musiałek P, Piwowarska W, Nessler J, Buszman P, Grajek S, Breborowicz P, Majka M, Ratajczak MZ, REGENT Investigators (2009) Intracoronary infusion of bone marrow-derived selected CD34+ CXCR4+ cells and non-selected mononuclear cells in patients with acute STEMI and reduced left ventricular ejection fraction: results of randomized, multicentre Myocardial Regeneration by Intracoronary Infusion of Selected Population of Stem Cells in Acute Myocardial Infarction (REGENT) Trial. Eur Heart J 30:1313–1321
Yeh YC, Wei HJ, Lee WY, Yu CL, Chang Y, Hsu LW, Chung MF, Tsai MS, Hwang SM, Sung HW (2010) Cellular cardiomyoplasty with human amniotic fluid stem cells: in vitro and in vivo studies. Tissue Eng Part A 16:1925–1936
Acknowledgments
The authors wish to thank Yajun Guo, Sheng hou, Xin Tong, Lei Zheng and Liangliang Wu for expert technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Jing Bai and Yiru Wang have the same contribution to this manuscript.
Rights and permissions
About this article
Cite this article
Bai, J., Wang, Y., Liu, L. et al. Human amniotic fluid-derived c-kit+ and c-kit− stem cells: growth characteristics and some differentiation potential capacities comparison. Cytotechnology 64, 577–589 (2012). https://doi.org/10.1007/s10616-012-9441-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-012-9441-6