
Abstract
Drosophila melanogaster Schneider 2 (S2) cells have been increasingly used as a suitable expression system for the production of different recombinant proteins, and the employment of bioreactors for large-scale culture is an important tool for this purpose. In this work, Drosophila S2 cells producing the rabies virus glycoprotein RVGP were cultivated in bioreactor, employing a serum-free medium, aiming an improvement in cell growth and in glycoprotein production. To overcome cell growth limitation commonly observed in stirred flasks, different experiments in bioreactor were performed, in which some system modifications were carried out to attain the desired goal. The study showed that this cell line is considerably sensitive to hydrodynamic forces, and a high cell density (about 16.0 × 106 cells mL−1) was only obtained when Pluronic F68® percentage was increased to 0.6% (w/v). Despite ammonium concentration affected RVGP production, and also cell growth, an elevated amount of the target protein was obtained, attaining 563 ng 10−7 cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Animal cell culture to obtain bioproducts has started in the 1950s with the production of vaccines for animals and humans, but its employment in large scale has become more usual since the 1980s due to recombinant DNA and hybridoma technologies (Augusto and Oliveira 2001). Herewith, improvement in the employed methodologies was necessary and the particular features of animal cells were so considered.
In this sense, the bioreactors used for culture of animal cells should be gently aerated and agitated, as high rates can cause shear injuries for cells. Environmental conditions should be homogeneous and well-controlled. Agitated tank bioreactors are the most frequently used since they provide adequate process control and are easy to scale up. Therefore, bioreactors design for animal cells include round-bottom geometry, suitable impellers and the prevention of vortex areas, which could cause shear damages (van der Pol and Tramper 1998).
In large-scale culture, protecting agents against hydrodynamic forces are commonly added to culture medium formulations, mainly to serum-free media, to avoid cell injuries. Commonly used additives include some polymers, such as Pluronic F68®, methylcellulose and polyethylene glycol, and fetal bovine serum. Among polymers, Pluronic F68®, a non-ionic surfactant, is the most employed for insect and mammalian cells (Palomares et al. 2000). Its utilization is related to higher cell survival and concentration, mainly in cultures performed in serum-free media. Nevertheless, the action mechanism of this additive is still debated (Palomares et al. 2000).
Some authors suggest that, in aerated cultures, Pluronic F68® presence minimizes cell-bubble adherence, decreasing the damages that occur when cells are carried to the liquid surface. Other proposed mechanisms include foam layer stabilization in the top of bioreactor and liquid film slower draining during bubble rupture, as well as reduction in membrane fluidity after association to cell membrane (Chisti 2000). This surfactant also presents effects in non-aerated cultures, in which cell damage occur due to vortex presence, reducing the tendency of cells adherence to the new surface (van der Pol and Tramper 1998; Palomares et al. 2000).
Dipteran’s embryonic cells such as Drosophila melanogaster Schneider 2 (S2) cells are being successfully employed in recombinant protein expression (Deml et al. 1999a, b), since they present some advantages as fast growth, non-requirement of pH control, and use of culture media that do not require expensive additives. Besides that, the Drosophila expression system is a non-lytic insect expression system that uses simple plasmid vectors. Biological products already expressed by these cells include antibodies (Kirkpatrick et al. 1995), receptors (Millar et al. 1995; Tota et al. 1995; Van den Broeck et al. 1995; Torfs et al. 2000; Perret et al. 2003), enzymes (Gibson et al. 1993; Banks et al. 2003), tumor inhibitors (Valle et al. 2001; Jeon et al. 2003), growth factors (Lee et al. 2000) and viral antigens (Deml et al. 1999a; Bachmann et al. 2004). Nevertheless, literature is still scarce with regard to metabolism and culture of these cells, with few previous works dealing specifically with recombinant rabies virus glycoprotein production through these cells (Yokomizo et al. 2007; Galesi et al. 2007).
In this study, Drosophila melanogaster Schneider 2 cells producing the rabies virus glycoprotein (RVGP) were cultured in bioreactor employing a serum-free culture medium. Some modifications in system were performed to bring cell behavior in bioreactor close to that observed in agitated flasks. Besides that, glycoprotein expression was evaluated considering the influence of cell growth, pH, lactate and ammonium production.
Materials and methods
Cell line and culture media
The transfected insect cell line S2AcRVGP2 producing the G glycoprotein from rabies virus was employed (Yokomizo et al. 2007). Cells were maintained in a serum-free TC100-based medium (Galesi et al. 2007), formulated with TC100 medium (Cultilab, Campinas, Brazil) containing 3 g L−1 of yeastolate and 1% (v/v) of a lipid emulsion (Gibco, Grand Island, USA), 10 g L−1 of glucose, 3.5 g L−1 of glutamine and 0.1% (m/v) of Pluronic F68® (Sigma Chemical Co., St. Louis, USA).
Inoculum preparation
For each bioreactor run, a vial of cells were thawed and cultivated in the previously described culture medium in 100 mL shake flasks (working volume of 20 mL) and incubated in a rotary shaker, at 100 rpm, and 28 °C, with subcultures occurring every 3–4 days. Cells with 25–30 passages in the culture medium, and at exponential growth phase, were used as inoculum in all experiments. Before inoculation in bioreactors, cells were centrifuged at 320 g for 5 min and the metabolized medium was discharged.
Bioreactor runs
Bioreactor runs were performed at a 2 L Biostat MD system (B. Braun, Germany), (1.5 L of working volume). Aeration was realized through a silicone tube (Si-Schlauch, Germany), with a diameter of 3.00 ± 0.20 mm and a wall thickness of 0.35 ± 0.15 mm. Total tube length was 5 m, but only 3.5 m was submersed. Temperature was controlled at 28 °C. Five runs were performed in distinct conditions aiming at adequate cell growth, as described in Table 1.
In the first two runs, oxygen from atmospheric air was supplied through an odontological compressor (Barionkar, EL 20750 model, Brazil), and its concentration was controlled feeding the system with ultra-pure nitrogen and ultra-pure oxygen (White Martins, Brazil). Two Rushton turbines were employed for stirring, providing radial flow in bioreactor. In the third run, medicinal synthetic air (Air Liquid, Brazil) substituted the odontological compressor, and mixing was performed with two pitched blade turbines with two paddles each (45° angle), providing axial flow. The forth and fifth runs were carried out with the same system, but increasing the Pluronic F68® percentage to 0.3 and 0.6% (w/v), respectively.
All experiments were performed with an inoculum concentration of 7.5 × 105 viable cells mL−1.
Analytical methods
Cell concentration was determined by optical microscopy through a Neubauer hemocytometer and viability by the trypan blue dye exclusion method (Freshney 1994). Glucose and lactate were determined by High Performance Liquid Chromatography—HPLC (Waters, EUA), utilizing the columns SC1011 and SH1011 (Shodex, EUA), respectively. Ammonium was determined by a specific electrode (Orion 9512, EUA). Glutamine concentration was determined enzymatically with a 2700 YSI Biochemical Analyser (Yellow Spring Instruments, USA).
The target protein (RVGP), a transmembrane protein, was recovered from the cells through centrifugation of sample aliquots containing 106 cells at 580 g for 5 min; the supernatant was discarded and the cell pellet was disrupted by using a lysis buffer (25 mM Tris, 25 mM NaCl, 5 mM MgCl2) at pH 7.4 (Astray et al. 2008). After homogenization, the samples were kept at 4 °C for 1 h, with periodical mixing. Following the lysis process, samples were centrifuged for 5 min at 10,000 g for cell debris removal and the supernatants were analyzed. The target product concentration was measured employing a rabies glycoprotein ELISA kit (Pasteur Institute, France), with monoclonal antibodies specific to the bioactive trimeric form of the glycoprotein, and the RVGP cell content was estimated according to Eq. 1:
where RVGP is glycoprotein concentration, RVGP X is the glycoprotein cell content, X T is total cell concentration and t refers to time.
Results and discussion
With the purpose of evaluating S2AcRVGP2 cells growth in bioreactor, runs were performed employing a serum-free medium. Glucose consumption, lactate and ammonium productions were also evaluated. Results are presented in Figs. 1 and 2, and Table 2.

Cell growth (a) and viability (b) for runs 1 (●), 2 (■), 3 (△), 4 (◊) and 5 (□)

Glucose (a), glutamine (b), lactate (c) and NH +4 (d) profiles for runs 1 (●), 2 (■), 3 (△), 4 (◊) and 5 (□)
In the first run (Fig. 1a, Table 2), cells presented a lag phase of about 20 h and, after that, culture entered an exponential growth phase, with a maximum specific cell growth of 0.018 h−1, attaining a maximum cell concentration of 4.6 × 106 viable cells per mL. Nevertheless, this growth rate and cell concentration were inferior to that commonly obtained in agitated flasks (around 0.04 h−1 and 10 × 106 cells mL−1), and could be attributed to hydrodynamic forces caused by the high stirring rate employed (150 rpm), since cell debris were observed in the supernatant during the whole experiment. The reduction of the stirring rate to 100 rpm was performed in the second run, and it was observed that maximum cell concentration and growth rate were similar to those obtained in the previous run (5.0 × 106 cells mL−1 and 0.021 h−1, respectively) and cell viability decreased after 72–96 h (Fig. 1b), indicating that cells were under stress in such conditions. Apparently, the stirring rate decrease did not result on hydrodynamic forces reduction and, consequently, did not induce significant differences in cell behavior. However, these runs have allowed verifying that experimental errors are very low, since the curves obtained are quite similar.
In an attempt to minimize the differences observed between agitated flasks and bioreactor results, other modifications were made in the system (run 3). The radial flow impeller was substituted for an axial flow impeller, which is more adequate for cell culture since it provides less hydrodynamic forces. The stirring rate was also reduced and dissolved oxygen concentration was controlled at a lower value, closer to the values achieved in agitated flasks. In that system, O2 concentration is maintained at low levels because flasks are opened only once a day for sampling. Medicinal synthetic air was also employed to eliminate the possibility of toxicity due to the potential presence of toxic compounds, since atmospheric air was being used.
Despite all these modifications, cell growth was not superior to that obtained in the other two runs. Cells presented a lag phase duration of about 15 h, maximum cell concentration was 5.5 × 106 cells mL−1 (Fig. 1a), and maximum cell growth rate was 0.024 h−1 (Table 2). The stirring rate was increased during the culture period because pellet formation was observed at the bioreactor bottom potentially due to insufficient mixing.
The next attempt to overcome the lower growth of S2AcRVGP2 cells in the bioreactor consisted of increasing Pluronic F68® percentage, aiming to avoid limitations related to hydrodynamic forces. For this purpose, a previous experiment was performed in stirred flasks to assess the Pluronic F68® concentration upper limit that did not cause toxic effects to the cells. Despite the recommended concentration range of this surfactant to animal cells is from 0.05 to 0.3% (w/v), the range tested in this work was expanded, going from 0.1 to 0.6% (w/v), and no significant differences in cell growth rates were observed between the different cultures (data not shown). However, supplementation with values above 0.4% caused cell aggregation, which could interfere in nutrient transport, metabolite elimination and RVGP expression. Therefore, 0.3% of Pluronic F68® was added to the culture medium and another run was performed in bioreactor. In this situation, cell growth was higher than in the previous runs (Fig. 1a), achieving a final cell concentration closer to that observed in agitated flasks. No lag phase was observed, cell concentration attained 9.0 × 106 cells mL−1 and maximum specific cell growth rate was 0.033 h−1.
Despite restrictions about the use of Pluronic F68® percentages over 0.3%, a last effort was made to improve S2AcRVGP2 cell growth in bioreactor, increasing Pluronic F68® percentage to 0.6%. Figure 1a shows that the maximum cell concentration was around 16.1 × 106 cells mL−1, a value considerably higher than those observed in the previous runs and than that verified in the control experiment performed in an agitated flask (data not shown).
The lower growth in shaken flasks could be attributed to oxygen limitation, since in this culture system, air exchange occurs only with flask opening, while in bioreactor this condition was controlled. Swiech et al. (2008) showed that recombinant Drosophila melanogaster Schneider 2 cells present growth limitation, but not immediate death, due to oxygen deprivation in Schott-flask assays, with cells entering in stationary growth phase and remaining in it for long periods with high viabilities even at very low oxygen concentrations. The cells cultivated in bioreactor presented a lag phase of approximately 30 h, what could be credited to the low inoculum concentration (particularly in this run, an initial cell concentration of 4.8 × 105 cells mL−1 was practiced, a value 28% lower than the one employed in the remaining experiments). The higher Pluronic F68® concentration could also explain this behavior, since the cells were not previously adapted to this condition. Maximum specific cell growth rate was slightly lower than that observed in run 4, maybe due to the high Pluronic F68® concentration, although no differences were verified in small scale. These results show that probably the cells were not growing in large scale due to the higher hydrodynamic forces in bioreactor when compared to the agitated flasks, condition that was overcome with the addition of a higher concentration of Pluronic F68®.
Cell viability was up to 90% in all experiments (Fig. 1b), with exception of runs number 1 and 2, due to the higher agitation rate and type of impeller used.
Glucose consumption was similar in all runs (Fig. 2a). It was not the limiting substrate, since it was not totally consumed (except in run 5) and when cells reduced the growth rate, a significant amount of this substrate was still present in culture media. Glutamine analysis in experiments 2, 4 and 5 (Fig. 2b) shows that this amino acid was also not limiting, since at least 0.5 g L−1 of glutamine were present in culture medium when cells had their growth rate reduced. According to Bovo et al. (2008), Drosophila melanogaster Schneider 2 cell growth is indeed limited by the absence of glutamine in culture medium, which determines the end of exponential growth phase.
Lactate was produced in runs 1, 2, 4 and 5 in low quantities, 0.13–0.31 g L−1, and it was not produced in run 3. Ammonium production attained values up to 80 mg L−1 in runs 1, 2, 4 and 5, and around 67 mg L−1 in run 3. According to Ikonomou et al. (2003), insect cells are not as sensible as mammalian cells to ammonium presence, since the addition of 10 mM of ammonium salts to culture medium did not affect Sf-9 and BTI-EAA cell lines. On the other hand, the presence of 30 mM of NH4Cl had a great impact on recombinant protein production by High-FiveTM, despite affecting cell growth only discreetly. Studies also show that alanine is generally produced when glucose and amino acids are in excess in culture medium as well as ammonium is formed when there is glucose limitation (Ikonomou et al. 2003). This was probably not the case in the present work, since ammonium was produced in the presence of high glucose concentrations. Consistent values of 50–60 mg L−1 of ammonium were detected at the end of exponential phase (time indicated in Table 2), what can be an indication of cell growth inhibition by ammonium.
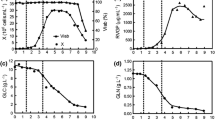
RVGP production was also measured, and the influence of cell growth, pH, lactate and ammonium production over glycoprotein expression were evaluated. As RVGP is a transmembrane protein, and S2AcRVGP2 cells have not a secretion signal, only cell lysates were analyzed. Figures 3 and 4 show that RVGP concentration increases with cell growth, diminishing after that. The highest concentration was obtained in run 2, in which 189 ng mL−1 of RVGP were produced in 116 h, while in run 3, the lowest levels of glycoprotein were observed. Regarding RVGP cell content, it is high or it increases in early culture, generally decreasing in the course of it. Increases in cell number did not result in correspondent augmentation in RVGP concentration, what maybe an evidence that glycoprotein production is more efficient in the early culture stage. RVGP cell content commonly presented high values in the runs, in a range of 300–400 ng 10−7 cells, attaining 563 ng 10−7 cells in run 5.

RVGP (□) and RVGP cell content ( ) (bar graphs), and viable cell concentration (▲), NH
+4
concentration (◊), lactate concentration (✶) and pH (○) for runs 1 (a), 2 (b) and 3 (c)
) (bar graphs), and viable cell concentration (▲), NH
+4
concentration (◊), lactate concentration (✶) and pH (○) for runs 1 (a), 2 (b) and 3 (c)

RVGP (□) and RVGP cell content () (bar graphs), and viable cell concentration (▲), NH
+4
concentration (◊), lactate concentration (✶) and pH (○) for runs 4 (a) and 5 (b)
RVGP cell content decreases whenever ammonium concentration reaches values around 45–55 mg L−1, suggesting that this by-product inhibits product synthesis, consistent to the previous observation that cell growth could also be inhibited by ammonium. For baculovirus expression system based on Trichoplusia ni cells, the literature reports that high ammonium concentrations reduces or even inhibits protein production (Yang et al. 1996; Ikonomou et al. 2003).
Since the RVGP undergoes structural changes in acidic pH, loosing its native shape (Gaudin et al. 1993; Gaudin 1997; Maillard and Gaudin 2002), pH and lactate concentration were monitored. In the five runs, only slight pH variations were observed, but the acidic culture medium pH could indeed have interfered with glycoprotein shape and also in its dosage, causing an underestimation of the actual produced RVGP concentration. An attempt of growing this cell line at a higher pH (6.8) was not successful, since the cells could not grow (data not shown). Although no significant changes were noted in pH during the runs, lactate production could have changed the internal cell pH, contributing to the loss of RVGP native structure and affecting protein quantification. Nevertheless, observing the results, it is not clear if lactate concentration played this role, since the quantity of this metabolite in the culture medium varies among runs when RVGP concentration decreased.
Concluding remarks
Despite being considerably sensitive to hydrodynamic forces, it is possible to cultivate S2AcRVGP2 cells in stirred bioreactors in serum-free medium if relatively high amounts of Pluronic F68® are added, achieving high levels of RVGP. Indications that ammonium interferes with glycoprotein production, as well as with cell growth were noted, however, the use of a fedbatch system, in which the gradual nutrient provision allows effective metabolite formation control, could overcome this limitation, turning the S2AcRVGP2 cells a potential platform for the production of a rabies virus vaccine.
References
Astray RM, Augusto EFP, Yokomizo AY, Pereira CA (2008) Analytical approach for extraction of recombinant membrane viral glycoprotein from stably transfected Drosophila melanogaster cells. Biotechnol J 3(1):98–103
Augusto EFP, Oliveira MS (2001) Processos com Células Animais. In: Biotecnologia Industrial, 1st edn, vol 3. Edgard Blücher, São Paulo
Bachmann AS, Corpuz G, Hareld WP, Wang G, Coller BA (2004) A simple method for the rapid purification of copia virus-like particles from Drosophila Schneider 2 cells. J Virol Methods 115:159–165
Banks DJ, Hua G, Adang M (2003) Cloning of a Heliothis virescens 110 kDa aminopeptidase N and expression in Drosophila S2 cells. Insect Biochem Mol 33:499–508
Bovo R, Galesi ALL, Jorge SAC, Piccoli RAM, Moraes AM, Pereira CA, Augusto EFP (2008) Kinetic response of a Drosophila melanogaster cell line to different medium formulations and culture conditions. Submitted to Cytotechnology
Chisti Y (2000) Animal-cell damage in sparged bioreactors. Trends Biotechnol 18:420–432
Deml L, Wolf H, Wagner R (1999a) High level expression of hepatitis B virus surface antigen in stably transfected Drosophila Schneider-2 cells. J Virol Methods 79:191–203
Deml L, Schirmbeck R, Reimann J Wolf H, Wagner R (1999b) Purification and characterization of hepatitis B virus surface antigen particles produced in Drosophila Schneider-2 cells. J Virol Methods 79:205–217
Freshney RI (1994) Culture of animal cells: a manual of basic technique, 3rd edn. Wiley-Liss, New York
Galesi ALL, Pereira CA, Moraes AM (2007) Culture of transgenic Drosophila melanogaster Schneider 2 cells in serum-free media based on TC100 basal medium. Biotechnol J 2(11):1399–1407
Gaudin Y (1997) Folding of rabies virus glycoprotein: epitope acquisition and interaction with endoplasmic reticulum chaperones. J Virol 71(5):3742–3750
Gaudin Y, Ruigrok RWH, Knossow M, Flamand A (1993) Low-pH conformational changes of rabies virus glycoprotein and their role in membrane fusion. J Virol 67(3):1365–1372
Gibson KR, Vanek PG, Kaloss WD, Collier GB, Connaughton JF, Angelichio M, Livi GP, Fleming PJ (1993) Expression of dopamine-β-hydroxylase in Drosophila Schneider 2 cells – evidence for a mechanism of membrane-binding other than uncleaved signal peptide. J Biol Chem 268:9490–9495
Ikonomou L, Schneider YJ, Agathos SN (2003) Insect cell culture for industrial production of recombinant proteins. Appl Microbiol Biotechnol 62:1–20
Jeon HK, Chang KH, Kim KI, Chung IK (2003) Functional expression of recombinant tumstatin in stably transformed Drosophila melanogaster S2 cells. Biotechnol Lett 25:185–189
Kirkpatrick RB, Ganguly S, Angelichio M, Griego S, Shatzman A, Silverman C, Rosenberg M (1995) Heavy chain dimers as well as complete antibodies are efficiently formed and secreted from Drosophila via a BiP-mediated pathway. J Biol Chem 270(34):19800–19805
Lee JM, Park JH, Park JO, Chang KH, Chung IS (2000) Expression of recombinant erythropoietin in stably transformed Drosophila melanogaster S2 cells. In Vitro Cell Dev Biol Anim 36:348–350
Maillard AP, Gaudin Y (2002) Rabies virus glycoprotein can fold in two alternative, antigenically distinct conformations depending on membrane-anchor type. J Gen Virol 83:1465–1476
Millar NS, Baylis HA, Reaper C, Bunting R, Mason WT, Sattelle DB (1995) Functional expression of a cloned Drosophila muscarinic acetylcholine receptor in a stable Drosophila cell line. J Exp Biol 198:1843–1850
Palomares LA, González M, Ramírez OT (2000) Evidence of Pluronic F68® direct interaction with insect cells: impact on shear protection, recombinant protein, and baculovirus production. Enzyme Microb Technol 26:324–331
Perret BG, Wagner R, Lecat S, Brillet K, Rabut G, Bucher B, Pattus F (2003) Expression of EGFP-amino-tagged human mu opioid receptor in Drosophila Schneider 2 cells: a potential expression system for large-scale production of g-protein coupled receptors. Protein Expr Purif 31:123–132
Swiech K, da Silva CS, Arantes MK, dos Santos AS, Astray RM, Pereira CA, Suazo CAT (2008) Characterization of growth and metabolism of Drosophila melanogaster cells transfected with the rabies-virus glycoprotein gene. Biotechnol Appl Biochem 49:41–49
Torfs H, Shariatmadari R, Guerrero F, Parmentier M, Poels J, Van Poyer W, Swinnen E, De Loof A, Akerman K, Van den Broeck J (2000) Characterization of a receptor for insect tachykinin-like peptide agonists by functional expression in a stable Drosophila Schneider 2 cell line. J Neurochem 74:2182–2189
Tota MR, Xu L, Sirotina A, Strader CD, Graziano MP (1995) Interaction of [fluorescein-Trp25]glucagon with the human glucagon receptor expressed in Drosophila Schneider-2 cells. J Biol Chem 270:26466–26472
Valle MA, Kester MB, Burns AL, Marx SJ, Spiegel AM, Shiloach J (2001) Production and purification of human menin from Drosophila melanogaster S2 cells using stirred tank reactor. Cytotechnology 35:127–135
van der Pol L, Tramper J (1998) Shear sensitivity of animal cells from a culture-medium perspective. Trends Biotechnol 16:323–328
Van den Broeck J, Vulsteke V, Huybrechts R, De Loof A (1995) Characterization of a cloned locust tyramine receptor cDNA by functional expression in permanently transformed Drosophila S2 cells. J Neurochem 64:2387–2395
Yang JD, Gecik P, Collins A, Czarnecki S, Hsu HH, Lasdun A, Sundaram R, Muthukumar G, Silberklang M (1996) Rational scale-up of a baculovirus-insect cell batch process based on medium nutritional depth. Biotechnol Bioeng 52:696–706
Yokomizo AY, Jorge SAC, Astray RM, Fernandes I, Filho ORG, Horton DSPQ, Tonso A, Pereira CA (2007) Rabies virus glycoprotein expression in Drosophila S2 cells. I. Functional recombinant protein in stable co-transfected cell line. Biotechnol J 2(1):102–109
Acknowledgements
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brasília, Brazil), Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP Grant Nos. 02/09482-3 and 03/00675-6, São Paulo, Brazil), Instituto Butantan (São Paulo, Brazil) and Instituto de Pesquisas Tecnológicas (São Paulo, Brazil).
Author information
Authors and Affiliations
Corresponding author
Additional information
Ângela M. Moraes is recipient of a CNPq fellowship.
Rights and permissions
About this article
Cite this article
Galesi, A.L.L., Aguiar, M.A., Astray, R.M. et al. Growth of recombinant Drosophila melanogaster Schneider 2 cells producing rabies virus glycoprotein in bioreactor employing serum-free medium. Cytotechnology 57, 73–81 (2008). https://doi.org/10.1007/s10616-008-9139-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-008-9139-y