
An unprecedented bis-indole 1, a novel indole alkaloid 2, and some known indole-related compounds 3–5 have been isolated from the sponge Plakortis sp. collected from Zampa in Okinawa. Their structures were elucidated by spectroscopic analysis. These metabolites showed cytotoxic activity against P388 leukemia and B16 melanoma cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
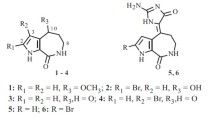
Marine organisms are rich sources of a variety of natural products possessing unique skeletons with many functional groups. Marine sponges have proved to be sources of bioactive compounds [1, 2]. Among them, indole alkaloids are commonly found in marine organisms and terrestrial plants as well as in microorganisms [3]. The bis-indole alkaloids belong to a class of marine natural products that may serve as a promising candidate for new drug leads. Some examples are staurosporine [4, 5], coscinamide [6], and chondriamide [7]. They exhibit a wide range of biological activities such as protein kinase inhibition [4, 5], anti-HIV [6], and cytotoxicities [7]. The sponge genus Plakortis usually produces major cyclic peroxide compounds [8], highly volatile ketone or acid [9], alkaloid [10], aromatic compounds [11], and fatty acids [12]. In the course of our studies on the bioactive molecules of marine organisms, we found two novel indole alkaloid compounds (1, 2) together with the known indole compounds penaresin (3), indolecarbaldehyde (4), and plakohypaphorine D (5) whose structures (for the new ones) and biological activities are described herein.

The sponge was extracted with MeOH exhaustively. The MeOH extract was then partitioned between EtOAc and H2O to give the cytotoxic EtOAc layer at 1 μg/mL, which was subjected to chromatographic separation to give compounds 1 (0.00012% wet weight), 2 (0.00004% wet weight), and the known compounds 3–5. Compound 1 was expected to have the molecular formula C21H16N2 as indicated by FAB and NMR data (15 degrees of unsaturation). This can be explained by the presence of two indole ring systems. One indole ring showed δ 7.51 (1H, br.s), 7.38 (1H, d, J = 8.0 Hz), 7.16 (1H, m), 7.12 (1H, m), and 7.86 (d, J = 7.3 Hz). Another indole ring system showed δ 7.38 (1H, d, J = 8.0 Hz), 7.12 (1H, m), 7.08 (1H, m), 7.66 (1H, d, J = 7.3 Hz), and 8.48 (1H, s). These two indole rings were connected by two trans double bonds. One bond showed δ 6.45 (1H, d, J = 16.1 Hz), 117 (d), 7.76 (1H, d, J = 16.1 Hz), and 136.5 (d). The other showed δ 7.01 (1H, d, J = 12 Hz), 129.5, 5.78 (1H, d, J = 12 Hz), and 116.6. Another structural feature was the presence of an ester at δ 174. The indole ring and the trans double bond were connected by the HMBC correlation between H-2/C-10, H-2/C-11, and H-11/C-3, and another indole ring system was connected to another trans double bond by the HMBC correlation between H-11′/C-3′, H-11′/C-8′, H-10′/C-3′, and H-10′/C-2′. The carbon ester C-12′ was connected to two indole rings by the HMBC between H-11/C-12′ and H-11′/C-12′.
The elemental composition of 2 was determined by HR-ESI-MS to be C11H8N2O. A set of indole signals was observed at δ 7.91 (1H, br.s), 7.42 (d, J = 7.9 Hz), 7.18 (1H, m), 7.15 (1H, m), and 8.09 (d, J = 7.9 Hz). The presence of amide and alkyne was deduced from HR-ESI-MS data to give nine degrees of unsaturation. The scarcity of the sample precluded us from conducting the 13C NMR for a more rigorous assignment of the alkyne and amide functional groups. The whole structure of the compound was assigned as in 2.
The other fractions in the EtOAc layer showing cytotoxic activity against both P388 and B16 cells at a concentration of 1 μg/mL were purified by chromatography to give the known compounds 3–4. The structure of penaresin (3) and indolecarbaldehyde (4) was assigned on the basis of their NMR and mass spectra data. In addition, a fraction from the water layer of Plakortis sp. gave a cytotoxic component, which was further purified as plakohypaporine D (5). The cytotoxic activities of all compounds were evaluated using P388 and B16 cells. Compound 1 was inactive against both P388 and B16 cells even at 100 μg/mL, while 2 showed cytotoxicity against P388 at 1 μg /mL (IC50 0.6 μg/mL) and B16 cells at 100 μg/mL. Compounds 3, 4, and 5 were active against P388 cells: IC50 5 μg/mL, <0.1 μg/mL, and 3.2 μg /mL, respectively.
A number of indole alkaloid molecules derived from marine sources have been reported [13]; however, the presence of the enol ester found in 1 and the alkyne coupled with indole and amide functional groups in 2 has been rarely reported. Considering that metabolites derived from this sponge in minor quantities showed structural similarity with those from bacteria, the indole compounds (1–5) were assumed to be a product of symbiotic microorganisms. A series of cyclic peroxides was also isolated as major compounds in this sponge.
Experimental
General. 1D and 2D NMR spectra were obtained on a JEOL Delta 600 spectrometer at 800 MHz for 1H and 150 and 200 MHz for 13C spectra, respectively. CD3OD was used as NMR solvent and referenced to δH 3.31 and δC 49.0. Chemical shifts and coupling constants are given as ppm and Hz. ESI-MS spectra were measured on an ESI-TOF-MS QSTAR Pulsar MS, while FAB-MS spectra were determined on a JEOL JMS-L2000 spectrometer operating in the positive FAB mode (NBA as a matrix). HPLC separations were carried out on a JASCO PU-980 Intelligent pump equipped with a UV-970 Intelligent UV-Vis detector and refractive index detector. Reversed-phase silica gel (250 × 20 mm, 250 × 10 mm Develosil) was used for HPLC. Analytical TLC was performed on commercially available silica gel 60 F254 plates and monitored with anisaldehyde and a UV lamp at 254 nm. The microtube NMR used for NMR measurement was from Shigemi.
Animal Material. The sponge, Plakortis sp., was collected at 30 m depth off Zampa, Okinawa, Japan. The sponge was taken from an overhung cave and kept frozen until extraction.
Extraction and Isolation. The sponge (wet, 250 g) was extracted with MeOH exhaustively. The resulting residue (20.90 g) was partitioned between EtOAc and water, and the organic layer was concentrated to give 2.41 g of the extract. It was then separated on a VLC silica gel chromatograph with stepwise elution using n-hexane, n-hexane–EtOAc (1:1), EtOAc, and MeOH to give a total of four fractions. Fraction 2 (1.65 g) eluted with n-hexane–EtOAc (1:1), which showed activity against P388 leukemia and B16 melanoma cells, was then purified on an ODS gel chromatograph with stepwise elution using 20, 40, 60, and 80% aqueous MeOH and 100% MeOH to give three major subfractions. The active second subfraction was purified by ODS HPLC with isocratic elution by 30% aqueous MeCN for 40 min at a flow rate of 1 mL/min to give nine subfractions. Subfraction 3 was identified as compound 2 (0.1 mg). Subfraction 4 was identified as indolecarbaldehyde 4 (1.3 mg). Subfraction 6 was further purified by ODS HPLC with 35% aqueous MeCN for 15 min to give penaresin 3 (1.0 mg) and the novel bis-indole alkaloid 1 (0.3 mg). The water layer from the partition between EtOAc and water was further chromatographed using TSK Gel 3000S with stepwise elution by H2O, 25, 50, and 75% aqueous EtOH, and 100% EtOH. The fraction showing strong activity against P388 cells (0.1 μg/mL) was then purified using open column ODS and ODS HPLC (MeOH–H2O) to give the polar compound, plakohypaphorine D (5) (0.5 mg).
Compound 1. Clear colorless oil. 1H NMR (CD3OD, δ, ppm, J/Hz): 7.76 (1H, d, J = 16.1, H-11), 6.45 (1H, d, J = 16.1, H-10), 7.39 (1H, d, J = 8.0, H-7), 7.16 (1H, m, H-6), 7.12 (1H, m, H-5), 7.86 (1H, d, J = 7.3, H-4), 7.51 (1H, br.s, H-2), 7.01 (1H, d, J = 12, H-11′), 5.78 (1H, d, J = 12, H-10′), 7.38 (1H, d, J = 8.0, H-7′), 7.12 (1H, m, H-6′), 7.08 (1H, m, H-5′), 7.66 (1H, d, J = 7.3, H-4′), 8.48 (1H, s, H-2′). 13C NMR (CD3OD, δ, ppm): 129.1 (d, C-2), 113.5 (s, C-3), 120.1 (d, C-4), 120.7 (d, C-5), 122.4 (d, C-6), 111.9 (d, C-7), 126.2 (s, C-8), 138.5 (d, C-9), 117.0 (d, C-10), 136.5 (d, C-11), 174 (s, C-12, 12′), 129.2 (d, C-2′), 111.6 (s, C-3′), 117.5 (d, C-4′), 119.8 (d, C-5′), 120.7 (d, C-6′), 112.0 (d, C-7′), 128.6 (s, C-8′), 136.4 (s, C-9′), 116.6 (d, C-10′), 129.5 (C-11′), 174.0 (C-12′). It was not possible to obtain peaks for high accurate masses by high-resolution FAB because of the nature of the compound [13], although at low resolution such a mass spectrum showed a strong peak at m/z 329.2 [M + H]+.
Compound 2. Clear colorless oil. 1H NMR (CD3OD, δ, ppm, J/Hz): 7.91 (1H, br.s, H-2), 7.42 (1H, d, J = 7.9, H-7), 7.18 (1H, m, H-6, 5), 7.15 (1H, m, H-5, 6), 8.09 (1H, d, J = 7.9, H-4). HR-ESI-MS obsd [M + H]+ m/z 185.1177 calcd for C11H9N2O, 185.0637.
Compounds 3–5. Compound 3 is a clear colorless oil. 1H NMR data were compared with the literature [14]. HR-ESI-MS obsd [M + H]+ m/z 188.0689, calcd for C11H10NO2, 188.0633. Compound 4 is a clear colorless oil. 1H NMR data were compared with the literature [15]. HR-ESI-MS obsd [M + H]+ m/z 146.0575, calcd for C9H8NO 146.0606. Compound 5 is a clear colorless oil. 1H NMR data were compared with the literature [16]. HR-ESI-MS obsd [M + Na]+ m/z 520.9187, calcd for C14H16I2N2O2Na, 520.9199.
Cytotoxic Activities. Growing cells of murine P388 lymphocytic leukemia or B16 melanoma were suspended in RPMI-1640 medium containing 10% fetal bovine serum, 5 μM 2-hydroxyethyl disulfide, and kanamycin (100 μg/mL) at 2 × 104 cells/mL, and samples dissolved in acetone were added. The cells were incubated at 37°C for 4 days in a CO2 incubator with a humidified atmosphere containing 5% CO2. The cell numbers were counted by the MTT method [17]. The IC50 value (concentration required for 50% inhibition of cell growth) was determined using the growth curve.
References
D. Uemura, Chem. Rec., 6, 235 (2006).
T. Higa, J. Tanaka, A. Kitamura, T. Koyama, M. Takahashi, and T. Uchida, Pure Appl. Chem., 66, 2227 (1994).
M. Somei and F. Yamada, Nat. Prod. Rep., 22, 73 (2005).
S. Omura, Y. Iwai, A. Hirano, A. Nakagawa, J. Awaya, H. Tsuchiya, Y. Takahashi, and R. Masuma, J. Antibiot., 30, 275 (1977).
N. Funaro, H. Takayanagi, Y. Konda, Y. Toda, Y. Harigaya, and S. Omura, Tetrahedron Lett., 35, 1251 (1994).
H. R. Bokesch, L. K. Pannel, T. C. McKee, and M. Boyd, Tetrahedron Lett., 41, 6305 (2000).
J. A. Palermo, P. B. Flower, and A. M. Seldes, Tetrahedron Lett., 33, 3097 (1992).
S. Sakemi, T. Higa, U. Anthoni, and C. Christophersen, Tetrahedron, 43, 263 (1987).
D. J. Faulkner and B. N. Ravi, Tetrahedron Lett., 21, 23 (1980).
W. D. Inman, M. O_Neill-Johnson, and P. Crews, J. Am. Chem. Soc., 112, 1 (1990).
B. N. Ravi, R. W. Amstrong, and D. J. Faulkner, J. Org. Chem., 44, 3109 (1979).
S. Tsukamoto, S. Takeuchi, M. Ishibashi, and J. Kobayashi, J. Org. Chem, 57, 5255 (1992).
R. Watanadilok, P. Sonchaeng, A. Kijjoa, A. M. Damas, L. Gales, A. M. S. Silva, and W. Herz, J. Nat. Prod., 64, 1056 (2001).
J. Kobayashi, J. F. Cheng, S. Yamamura, T. Sasaki, and Y. Ohizumi, Heterocycles, 31, 2205 (1990).
J. H. Cardellina, D. Nigh, and B. C. van Wagenen, J. Nat. Prod., 49, 1065 (1986).
F. Borrelli, C. Campagnuolo, R. Capasso, E. Fattorusso, and T. Scafati, Eur. J. Org. Chem., 2004, 3227 (2004).
M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski, D. L. Fine, B. J. Abbot, R. H. Shoemaker, and M. R. Boyd, Cancer Res., 48, 589 (1988).
Acknowledgment
This work was supported in part by Grants-in-Aid for Scientific Research (No. 21221009) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 6, November–December, 2015, pp. 973–975.
Rights and permissions
About this article

Cite this article
Hanif, N., Yamada, K., Kitamura, M. et al. New Indole Alkaloids from the Sponge Plakortis sp.. Chem Nat Compd 51, 1130–1133 (2015). https://doi.org/10.1007/s10600-015-1508-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-015-1508-0