
Bioassay-guided isolation studies on the methanolic extract of the leaves of Bombax ceiba employing DPPH antioxidant assay led to the isolation of a new xanthone C-glucoside, shamimoside (2), along with three known constituents, mangiferin (1), stigma-5-en-3-O-β-glucoside, and β-amyrin. The structure of shamimoside has been elucidated through extensive spectroscopic methods, including 1D and 2D NMR experiments, as 4-C-β-D-glucopyranosyl-1,3,6,8-tetrahydroxy-7-O-(p-hydroxybenzoyl)-9H-xanthen-9-one (2). It is the first naturally occurring xanthone containing a benzoate moiety directly attached to an aromatic ring. Polar extracts and fractions demonstrated better antioxidant activity, and 1 was found to be more potent than 2 in this assay.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bombax ceiba L. (B. malabaricum DC.), commonly known as Simbal and red silk cotton tree, belongs to the family Bombacaceae. Systematic pharmacological work was also done on it, e.g., determination of lipase activity of seeds [1], evaluation of hypotensive and toxicological effect of stem bark [2], hypoglycemic, hypotensive, and toxicological effect of leaf extract [3], and assessment of antiangiogenic [4] and anti-inflammatory activity [5]. B. ceiba is a reservoir of a wide range of natural products with diverse structure, of which the bioactive xanthone C-glycoside mangiferin (1) is a predominant constituent of its leaves. It can be obtained directly from the methanolic extract in substantial amounts [6–8]. Different classes of compounds that have been isolated from B. cieba included triterpenes, steroids, sesquiterpenoids, polysaccharides, anthocyanins, flavonoids, long-chain ferulic acid ester, and norneolignans [9–17].
Xanthones are secondary metabolites commonly occurring in a few higher plant families, in fungi and lichen [18]. Mangiferin (1), a glucosylxanthone, was the first xanthone to be investigated for pharmacological purposes [19].

In the course of our ongoing research on the chemistry and biological activity of local medicinal plants, a methanol-soluble portion of the MeOH extract of B. ceiba leaves was found to have significant antioxidant activity in DPPH assay. This extract was fractionated and purified by repeated chromatography employing the bioassay, which led to the isolation of a new antioxidant xanthone benzoate, shamimoside (2), along with two known constituents, stigma-5-en-3-O-β-glucoside [20] and β-amyrin [21]. From the methanol-insoluble portion, the active antioxidant principle mangiferin (1) has been obtained. During the isolation studies it was observed that the green parts of the buds and flowers (calyces) also contained mangiferin (1) as the major xanthone.
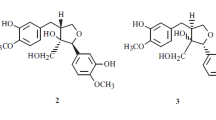
Shamimoside (2), obtained as a pale yellow powder, has the formula C26H22O14 (MW = 558), derived by the combined applications of FAB-MS (+ve), and 1H and 13C NMR data (Table 1), while its EI-MS spectrum showed no molecular ion peak, pointing to its glycosidic nature. The IR spectrum displayed hydroxyl and chelated carbonyl absorptions at 3440 and 1651 cm–1, respectively, the ester carbonyl and olefinic bond gave rise to absorptions at 1720 and 1631 cm–1, respectively, and a band at 1582 cm–1 was indicative of the aromatic system. Its UV spectrum showed maxima at 244, 265, 271, 340, and 365 nm, which are characteristic of 1,3,6,7,8-pentaoxygenated xanthone [22]. The 13C NMR spectra (DEPT) showed that the 26 carbons of the molecule are present as one methylene, five methine, six sp2 CH, and 14 sp2 quaternary carbons. The molecular formula showed 16 double-bond equivalence, indicating its highly aromatic nature. Its 1H NMR spectrum in CD3OD (Table 1) showed the presence of a sugar moiety by an anomeric proton doublet at δ 4.98 (J = 9.9 Hz, H-1'), a one-proton triplet at δ 4.09 (J = 9.9, H-2'), and a multiplet with integration of five protons at δ 3.21–3.83 for the remaining five protons (H-3' to H-6') of sugar. With the help of 1H–1H COSY-45°, DEPT spectrum, and one-bond heteronuclear (HMQC) experiment, the chemical shifts of sugar protons were assigned as δ 3.50 (H-3'), 3.21 (H-4'), 3.27 (H-5'), 3.83 (H-6a'), and 3.79 (H-6b'), which showed correlation with δ 80.2 (C-3'), 71.7 (C-4'), 78.1 (C-5'), and 62.7 (C-6'), respectively (Table 1).
These spectral data agreed with the existence of a β-linked D-glucopyranose moiety in the molecule, while the appearance of anomeric carbon signal at high field at δ 75.3 in the 13C NMR spectrum (Table 1) and the large 1H–1H coupling constant (J = 9.9 Hz) of the anomeric proton showed the linkage of sugar to the aromatic ring through carbon and not via oxygen atom [23]. Resistance to acid hydrolysis with HCl proved that shamimoside is C-glycoside in nature with a xanthone skeleton [23]. The high-frequency region of the 1H NMR spectrum showed four resonances, of which two-one proton singlets observed at δ 6.26 and 6.59 were assigned to H-2 and H-5 of ring A and ring B of the xanthone core on the basis of their similarity to the signals observed for iso-mangiferin and related compounds [24]. The singlet at δ 6.26 for H-2 is very significant as its low-frequency value indicated that it flanks between the two hydroxyl groups as in iso-mangiferin [24]. The HMQC spectrum showed the one-bond correlation of δ 6.26 (H-2) with C-2 (δ 99.4), while the HMBC plot confirmed the assignment exhibiting the very important correlation of H-2 (δ 6.26) with δ 163.0 (C-1), 104.1 (C-9a), 164.7 (C-3), and 105.5 (C-4). The characteristic peak at δ 184.3 for the carbonyl carbon in the 13C NMR spectrum showed the presence of hydroxyl groups at C-1 and C-8 of the xanthone moiety, which are chelated in nature [25]. This observation was confirmed by running the 1H NMR spectrum in C5D5N, which showed two broad D2O exchangeable singlets at δ 13.82 and 14.01 for chelated hydroxy groups (Table 1). Furthermore, the 1H NMR spectrum of 2 in CD3OD also displayed two aromatic doublets at δ 7.96 (J = 8.5 Hz) and 6.93 (J = 8.5 Hz) for the p-hydroxybenzoate moiety, with the integration of two protons of each, and these were ascribed to H-2'', H-6'' and H-3'', H-5'', respectively, of the p-hydroxybenzoate ring. These chemical shifts are comparable with those reported for phenyl benzoate and phenyl 4-hydroxybenzoate in the literature [26]. All the 16 double-bond equivalence of the molecule were accounted for by the sugar and benzoate moieties and the xanthone core. Moreover, the 13C NMR spectrum supported the attachment of the electron-withdrawing p-hydroxybenzoyl moiety at C-7 in ring B as the δC value of C-7 shifted to low frequency, and of C-6 and C-8 appearing at high frequency relative to the reported δC value of ring B of the xanthone backbone [27]. The HMBC plot reaffirmed the assignment showing the correlation of H-5 (δ 6.59) with C-6 (δ 166.4) and C-8a (δ 105.2). Additionally, the electron impact mass spectrum (Scheme 1) substantiated the attachment of the p-hydroxybenzoyl moiety showing very important retro Diels-Alder (rDA) fragments at m/z 314 (C13H14O9, fragment c), 244 (C13H8O5), and 123 (C6H3O3, fragment e). The cross-peak in the NOESY plot showing the spatial interaction of H-5 with H-2'' and H-6'' further confirmed the location of the ester group at C-7. The linkage of the glucopyranosyl moiety with the aromatic ring was determined to be C-4 of the xanthone nucleus by the HMBC experiment and by comparing the value with reported data [23]. The site of the glycosidic bond with C-4 of the xanthone nucleus was also evident from the glycosidation effect which produced relatively high-frequency shifting of the C-4 as compared with its chemical shift when it is unsubstituted [23, 24].

The HMBC spectrum showed the long-range correlation of the anomeric proton (H-1') with 0 158.0 (C-4a), reaffirming the location of the sugar moiety. Exact 1H NMR assignment of shamimoside was made by a combination of 2D NMR experiments, which included COSY-45°, NOESY, HMQC, and HMBC sequences. Once the 1H NMR spectrum has been completely assigned, the protonated carbons were ascribed by using a one-bond heteronuclear correlation (HMQC) experiment. The assignment of 14 quaternary carbon resonances was accomplished by comparison with the reported data of similar compounds and through DEPT and HMBC spectra [23, 24]. On the basis of these data the structure of shamimoside was elucidated as 4-C-β-Dglucopyranosyl-1,3,6,8-tetrahydroxy-7-O-(p-hydroxybenzoyl)-9H-xanthen-9-one (2), which was corroborated by the diagnostic peaks in the EI mass spectrum at m/z 342 (C20H6O6, fragment a, 3%), 324 (C20H4O5, fragment b, 4%), 283 (C13H13O7, fragment d, 44%), and 123 (C6H3O3, fragment e, 17%) (Scheme 1). It is important to note that it is the first naturally occurring xanthone bearing a benzoate moiety directly attached to an aromatic ring. Biogenetically it is worth mentioning that the stem wood of B. ceiba contained p-hydroxybenzoic acid [28].
The present bioassay revealed that mangiferin demonstrated similar magnitude of activities as that of rutin (a standard antioxidant) as reflected by their IC50 values (5.80 ± 0.96 μg/mL versus 5.56 ± 0.33 μg/mL). These polyphenols have the catechol moiety in their structures. On the other hand, a significant decline in activity of 2 (IC50= 150 μg/mL) compared to 1 was observed, which may be due to the benzoylation at 7-OH. In another work [28], on fresh leaves of the plant a mixture of mangiferin and isomangiferin was obtained that showed less antioxidant activity (IC50 = 12 μg/mL) compared to mangiferin (1). It is important to note that (2) is more closely related to isomangiferin than to mangiferin.
The antioxidant effect of the extract, fractions, and pure compounds of the dried leaves of B. ceiba was evaluated using the DPPH assay, which revealed that the activity accumulated in the polar extracts and fractions. The petroleum ether extract and its methanol-soluble and methanol-insoluble fractions showed no antioxidant activity at 200 μg/mL dose (Table 2), while the ethyl acetate extract possessed weak antioxidant activity (IC50 = 200 μg/mL), and, upon its fractionation, activity appeared in the polar fraction. The methanolic extract of the leaves exhibited strong antioxidant properties in this assay. On its division into methanol-soluble and methanol-insoluble fractions, the activity appeared in the latter fraction which was identified as pure mangiferin (1) (5.8 ± 0.96 μg/mL), comparable to rutin (a standard antioxidant) [7]. As the methanol-soluble fraction also showed antioxidant potential (IC50 = 50 μg/mL), it was subjected to vacuum liquid chromatography (VLC), which afforded one pure compound, stigma-5-en-3-O-β-D-glucoside [20] showing negligible activity. Another fraction from VLC (12F) possessed moderate antioxidant activity (40 μg/mL). On its purification through FCC, it afforded a new xanthone shamimoside (2) showing IC50 = 150 μg/mL. Moreover, the 50% methanolic extract of the leaves also has antioxidant properties. During these studies, it was observed that activity increased with polarity of the extracts and fractions in B. ceiba leaves.
Experimental
The IR (in KBr disc) spectrum was recorded on a JASCO A 302 spectrophotometer, and the EI, FAB (+ve) and (-ve) mass spectra were measured on Finnigan MAT 112 and JMS HX-110 spectrometers. The 1H NMR spectra were run in CD3OD and C5D5N on a Bruker Aspect AM-400 spectrometer operating at 400 MHz, while 13C NMR spectra were recorded at 100 MHz. Purity of the compounds was checked on silica gel 60GF254 precoated cards (0.2 mm thickness), while for flash column chromatography (FCC) (Model Aldrich) silica gel 9385 (E. Merck) was used. For VLC, silica gel 60 GF254 was used.
Plant Material. The dried leaves of B. ceiba were collected in August 2003 from University of Karachi Campus. The plant was authenticated by Prof. Dr. Surraya Khatoon at the Department of Botany, University of Karachi, and a voucher specimen (No. 66854 KUH) was deposited in the same Department.
Extraction and Isolation. Dried and uncrushed leaves of B. ceiba (1.6 kg) collected from the University of Karachi Campus were extracted thrice with petroleum ether at room temperature. The petroleum ether extracts were combined and freed of the solvent in vacuo to give a residue (BCLD-PE, 31.67 g) which on treatment with methanol afforded methanol-soluble and methanol-insoluble fractions (BCLD-PEMI). The marc from petroleum ether extract was extracted with EtOAc twice, and the extracts were dried on sodium sulfate (anhydrous) and then charcoaled and filtered. The golden yellow filtrate thus obtained was freed of solvent in vacuo to a residue (BCLD-EA-C, 127.21g), while the charcoal bed was eluted with methanol–benzene (1:1) to afford the methanol–benzene eluate (BCLD-EA-MB). The marc left after E.A extraction was percolated with methanol twice, and the methanolic extracts were freed of solvent under reduced pressure to give a residue (BCLDM) which was treated with methanol to afford the methanol-soluble and methanol-insoluble (BCLDM-MI) fractions. The insoluble matter was washed with hot CH3OH, H2O, and EtOAc to furnish a yellowish green powder (BCLD-P, 11.57 g), which showed a single spot on TLC (silica gel, EtOAc–acetone–formic acid–H2O, 8:2:0.5:0.5, R f 0.23) and which was identified as mangiferin (1) [6]. The washings were combined with the methanol-soluble fraction and evaporated to give a residue (BCLDM-MM, 120.25 g). A portion of (BCLDM-MM, 80 g) was subjected to VLC (silica gel, petroleum ether, EtOAc, CH3OH, and H2O, in order of increasing polarity), affording 25 fractions. One of these fractions 12B (EtOAc–CH3OH, 9.0:1.0) showed a single iodine active spot on TLC and was identified as stigma-5-en-3-O-β-glucoside [20] (250 mg), while fraction 12C (EtOAc–CH3OH, 8.5:1.5, 360 mg) was subjected to FCC (silica gel, petroleum ether, EtOAc, CH3OH, and H2O, in order of increasing polarity by 2.5%), furnishing 90 fractions, of which fraction 43 (EtOAc–CH3OH, 9.5:0.5, 150 mg) showed a single spot on TLC (silica gel, EtOAc–CH3OH–H2O, 9.0:1.0:1.0, R f 0.55) and was characterized as a new xanthone derivative, shamimoside (2).
In another attempt, BCLDM-MM (36.05 g) was divided into 16 fractions through solvent-solvent separation (petroleum ether, EtOAc, CH3OH, and H2O soluble fractions). Fraction 14 (EtOAc–CH3OH, 1:1, 2.35 g) was subjected to FCC (silica gel, petroleum ether, EtOAc, CH3OH, and H2O, in order of increasing polarity), which gave 148 fractions. Fractions 15 (petroleum ether–EtOAc, 8.5:1.5, 75.0 mg) and 81 (EtOAc–CH3OH, 8.5:1.5, 120.2 mg) showed a single iodine and UV active spots on TLC and were characterized as -amyrin [21] (silica gel, petroleum ether–EtOAc, 9.5:0.5, R f 0.43) and xanthone derivative (2), respectively.
4-C-β-D-Glucopyranosyl-1,3,6,8-tetrahydroxy-7- O -( p -hydroxybenzoyl)-9 H -xanthen-9-one (2). Yellow powder. UV (MeOH, λmax, nm): 244, 265, 271, 340, 365. IR (CHCl3, νmax, cm–1): 3440, 1720, 1651, 1631, 1582, 1294, 1203, 1125, 1083; FAB-MS (+ve) m/z 559 (M+ + 1, C26H23O14). EI-MS (m/z, I rel, %): 342 (C20H6O6, fragment a, 3), 324 (C20H4O5, fragment b, 4), 314 (C13H14O9, fragment c, 4), 283 (C13H13O7, fragment d, 44), 123 (C6H3O3, fragment e, 17). 1H and 13C NMR, see Table 1.
DPPH Free Radical Scavenging Assay. This method is based on the scavenging activity of stable 1,1-diphenyl-2picrylhydrazyl (DPPH) free radicals [7]. The reaction mixture containing 30–200 μg/mL of the test samples and 300 fM DPPH ethanolic solution were left at room temperature for 30 min. Absorbance was measured at 517 nm on a UV-VIS spectrophotometer (Shimadzu UV-2200) and the percent inhibition after sample treatment was calculated.
Reference
M. W. Akhtar, H. Parveen, S. Kausar, and M. I. D. Chughtai, Pak. J. Biochem., 8, 77 (1975); Chem. Abstr., 85, 174292s (1976).
R. Saleem, S. I. Ahmed, Z. Faizi, S. Z. Rehman, M. Ali, and S. Faizi, Biol. Pharm. Bull., 26, 41 (2003).
R. Saleem, M. Ahmad, S. A. Hussain, A. M. Qazi, S. I. Ahmad, M. H. Qazi, M. Ali, S. Faizi, S. Akthar, and S. N. Husnain, Planta Med., 65, 331 (1999).
Y-J. You, N-H. Nam, Y. Kim, K-H. Bae, and B-Z Ahn, Phytother. Resear., 17, 341 (2003).
C. C. Lin, S. Y. Chen, J. M. Lin, and H. F. Chiu, Am. J. Chem. Med., 20, 135 (1992).
A. A. Shahat, R. A. Hassan, N. M. Nazif, S. V. Miert, L. Pieters, F. M. Hammuda, and A. J. Vlietinck, Planta Med., 69, 1066 (2003).
A. Dar, S. Faizi, S. Naqvi, T. Roome, S. Z. Rehman, M. Ali, S. Firdous, and S. T. Moin, Biol. Pharm. Bull., 28, 596 (2005).
S. Faizi, S. Z. Rehman, M. Ali, and A. Naz, Magn. Reson. Chem., 44, 838 (2006).
L. S. Puckhaber and R. D. Stipanovic, J. Nat. Prod., 64, 260 (2006).
A. A. Bell, R. D. Stipanovic, D. H. O’Brien, and P. A. Fryxell, Phytochemistry, 17, 1297 (1978).
A. V. B. Sankaram, N. S. Reddy, and J. N. Shoolery, Phytochemistry, 20, 1877 (1981).
K. Seeramulu, K. V. Rao, C. V. Rao, and D. Gunasekar, J. Asian Nat. Prod. Res., 3, 261 (2001).
G. D. Agrawal, S. A. I. Rizvi, P. C. Gupta, and J. D. Tewari, Planta Med., 21, 293 (1972).
G. S. Niranjan and P. C. Gupta, Planta Med., 24, 196 (1973).
J. S. Chauhan, M. Sultan, and S. K. Srivastava, Can. J. Chem., 58, 328 (1980).
P. Singh, D. K. Mewara, and M. C. Sharma, Nat. Prod. Commun., 3, 223 (2008).
J. Wu, X-H. Zhang, S-W. Zhang, and L-J. Xuan, Helv. Chem. Acta, 91, 136 (2008).
V. Peres, T. J. Nagem, and F. F. de Oliveira, Phytochemistry, 55, 683 (2000).
M. M. M. Pinto, M. E. Sousa, and M. S. J. Nascimento, Curr. Med. Chem., 12, 2517 (2005).
S. Faizi, M. Ali, R. Saleem, Irfanullah, and S. Bibi, Magn. Reson. Chem., 39, 399 (2001).
C. Soldi, M. G. Pizzolatt, A. P. Luiz, R. Marcon, F. C. Meotti, L. A. Mioto, and A. R. S. Santos, Biorg. Med. Chem., 16, 3377 (2008).
S. P. Gunasekera, S. Ramachandran, S. Selliah, and M. U. S. Sultanbawa, J. Chem. Soc. Perkin Trans. 1, 2447 (1975).
Y. Ikeya, K. Sugama, and M. Maruno, Chem. Pharm. Bull., 42, 2305 (1994).
T. Fujita, D-Y. Liu, S. Ueda, and Y. Takeda, Phytochemistry, 31, 3997 (1992).
S. Gil, P. Palanca, V. Sanz, and A. Tortayada, J. Nat. Prod., 53, 1198 (1990).
S-I. Sasaki, Asahi Research Center, Handbook of Proton-NMR Spectra and Data, Vol. 4, Academic Press, Inc. Tokyo, 1985, pp. 360–361.
H. Wada, Y. Shimizu, N. Tanaka, R. C. Cambie, and J. E. Braggins, Chem. Pharm. Bull., 43, 461 (1995).
S. Z. Rehman, Isolation of Bioactive Chemical Constituents from Bombax ceiba and Aegle marmelos, Ph.D. Dissertation, H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan, 2009.
Acknowledgment
One of the authors (Rehman S. Z.) is grateful to the Higher Education Commission Islamabad, Pakistan for financial support through the Merit Scholarship Scheme for Ph.D. Studies in Science and Technology (200 Scholarships).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2012, pp. 692–696.
Rights and permissions
About this article
Cite this article
Faizi, S., Zikr-ur-Rehman, S., Naz, A. et al. Bioassay-guided studies on Bombax ceiba leaf extract: isolation of shamimoside, a new antioxidant xanthone C-glucoside. Chem Nat Compd 48, 774–779 (2012). https://doi.org/10.1007/s10600-012-0379-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-012-0379-x