
Analysis of the reactivity indices shows the polar character of the (2+3) cycloaddition between (E)-3,3,3-tri-fluoro-1-nitroprop-1-ene and (Z)-N-methyl-C-phenylnitrone. This is confirmed by exploration of reaction paths by B3LYP/6-31G(d) algorithm. Although the transition complexes involved in the reaction mechanism are considerably asymmetric and polar, the reactions proceed via a one-step mechanism.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Zanda et al. [2] have found recently that the cycloaddition of (E)-3,3,3-trifluoro-1-nitroprop-1-ene (1) to (Z)-N-methyl-C-phenylnitrone (2) yields a mixture of (rel-3S,4R,5S)- (3) and (rel-3S,4S,5R)-2-methyl-4-nitro-3-phenyl-5-(3,3,3-trifluoromethyl)isoxazolidines (4) as the only reaction products.
The stereospecificity of reaction paths A and B suggests a concerted mechanism [3]. However, in the case of [4+2] π-electron cycloadditions involving strongly π-deficient alkenes, such as is obviously nitroalkene 1, a two-step zwitterionic mechanism [4-7] in the formation of products 3 and 4 may compete with the

concerted mechanism. The latter may occur stereospecifically if the rotation barrier (ΔG ≠ rot) within the zwitterionic intermediate is higher than the heterocyclic ring closure barrier (ΔG ≠ cycl), thus precluding the formation of the alternative products 5 and 6 [5]. It should be noted at this point that a two-step mechanism in the reaction between compounds 1 and 2 is more likely than in the case of diarylnitrone–nitroalkene (2+3) cycloaddition, because the global electrophilicity of compound 2 is lower than that of diarylnitrones [8]. Moreover, the nucleophilic oxygen atom in the CNO moiety of compound 2 is relatively weakly shielded. On the other hand, it is known that the presence of CF3 groups in the vicinity of the reaction centers may favor stabilization of zwitterionic intermediates [4].

To study the nature of the changes that occur in reactions 1+2→3 and 1+2→4, our investigation included: (i) analysis of electron interactions between reactant molecules with respect to the recently widely promoted [8-10] theory of reactivity indices and (ii) B3LYP/6-31G*(PCM) [11, 12] simulations of competing reaction pathways A and B.
The reactivity indices, that is, the electronic chemical potential (μ), global electrophilicity (ω), and static polarity (ΔN 0) of reactants 1 and 2, were calculated using the relationships [8-10]:
where μ = (E HOMO+E LUMO)/2 and η = E LUMO–E HOMO.
Reaction pathways were studied as we did before with respect to the cycloaddition reaction of (Z)-C,N-di-phenylnitrone to nitroethene [13], (E)-3,3,3-trichloro-1-nitropro-1-ene [14], and 2-nitroprop-1-ene [15]. In particular, for structure optimization of the reactants and the reaction products, the Berny algorithm [16] was applied. First-order saddle points were localized using the QST2 procedure. Stationary points were checked by vibrational frequency analyses to see whether they constituted minima or maxima on the potential energy surface. All transition structures showed a single imaginary frequency, whereas reactants, products, and pre-reaction complexes had none. The intrinsic reaction coordinate path was traced in order to check the energy profiles connecting each transition structure to the two associated minima of the proposed mechanism. For the optimized structures, the thermochemical data for temperature 298 K and pressure 1 atm were computed using vibrational analysis data. For the calculations of the solvent effect, the PCM algorithm of Tomasi et al. [12] was used.
Consistently with the previous [13, 15] conventions, in this paper the pre-reaction complexes are denoted as LM and the transition complexes as TS. They are distinguished by appending the letters A and B, depending on the reaction pathway.
By comparing μ values, it is seen that charge transfer in both reactions proceeds from nitrone 2 (μ = -0.1244 a.u.) to nitroalkene 1 (μ = -0.1711 a.u.). This is also confirmed by the ω values 1.35 and 3.24 eV, respectively. Because the electrophilicity difference (Δω) of reactants 1 and 2 is much higher than 1 eV, (2+3) cycloaddition reactions involving them can be regarded as polar processes [8]. The charge transferred in the reaction transition complex from the nitrone substructure to the nitroalkene substructure can be evaluated based on the ΔN 0 index. It is 0.26 e for both reactions. Therefore, μ, Δω, and ΔN 0 values confirm that the reactions tested may proceed through polar intermediates.
When the reactivity indices are known, the nature of interactions between the reactant molecules (Fig. 1) during cycloaddition can be predicted [8-10]. This, however, is insufficient to predict whether such reactions are concerted or they proceed, instead, through a stage of a zwitterionic intermediate as two-stage processes.

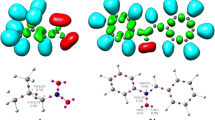
Views of (E)-3,3,3-trifluoro-1-nitroprop-1-ene (1) and (Z)-C-phenyl-N-methylnitrone (2) in toluene solution according to B3LYP/6-31G*(PCM) calculations.
Hence, the next stage of our study included B3LYP/6-31G*(PCM) simulations of actual reaction pathways A and B. We assumed that both reactions occurred in toluene. This solvent was used as the reaction medium also in earlier studies [2].
The calculations revealed a considerable similarity of energy profiles for both pathways (Fig. 2). In both cases, only one transition state (TS) preceded by the pre-reaction complex (LM) energy minimum was found between the enthalpic minima of the reactants 1+2 and corresponding cycloadducts 3 or 4. The attempts to find zwitterionic intermediates on pathways A and B were unsuccessful.

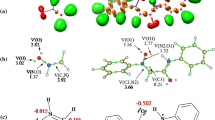
The reaction profiles for (2+3) cycloaddition reaction between nitroalkene 1 and nitrone 2 in toluene solution according to B3LYP/6-31G*(PCM) calculations.
The presence of LM A and LM B on the reaction pathways reduces the enthalpy of the reacting system by 3.6 and 6.2 kcal/mol, respectively, and its entropy – by as much as 31.4 and 29.2 cal/mol⋅K. No charge transfer is observed between the complex substructures (t ≈ 0.0 e). Therefore, they are not charge-transfer complexes [17, 18]. Both have features of orientation complexes [19], because the reaction centers are located in them identically, as subsequently observed in cycloadducts 3 and 4 (Fig. 3).

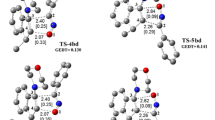
Views of LM and TS structures for (2+3) cycloaddition reaction between nitroalkene 1 and nitrone 2 in toluene solution according to B3LYP/6-31G*(PCM) calculations.
The formation of TS A and TS B coincides with increase in the system enthalpy of activation (ΔH ≠) by 9.2 and 8.2 kcal/mol, respectively, and decreased in the entropy of activation (ΔS ≠) by 49.6 and 48.8 cal/mol⋅K. High negative ΔS ≠ values suggest a concerted reaction mechanism. This is also confirmed by the geometric parameters of the complexes (Fig. 3, Table 1). New σ-bonds within them are formed simultaneously, even though the degree of their formation (l) varies (the C(5)–O(1) bond forms most rapidly). The corresponding length in both TS's is ~1.72 Å (l = 0.80), while the distance between reaction centers at C(3) and C(4) is more than 2.40 Å (l < 0.46). Therefore, the structures of TS A and TS B are largely asymmetric. This is confirmed by the Δl = l C(5)–O(1) - l C(3)–C(4) values (Table 1) being 0.38 and 0.34, respectively. The TS A and TS B complexes are distinctly polar. This is verified (Table 1) by their dipole moment values (μ D ) and the degree of charge transfer between substructures (t). However, the asymmetry of the complexes is not high enough to induce an ionic reaction.
Finally, it should be concluded that analysis of reactivity indices and results from exploration of reaction paths indicate polar character of the (2+3) cycloaddition between (E)-3,3,3-trifluoro-1-nitroprop-1-ene and (Z)-N-methyl-C-phenylnitrone. However, although the transition complexes involved in the reaction mechanism are considerably asymmetric and polar, the reactions still proceed via a one-step mechanism.
References
R. Jasiński, M. Mikulska, and A. Barański, Khim. Geterotsikl. Soedin., 857 (2013). [Chem. Heterocycl. Compd., 49, 802 (213)].
S. Bigotti, L. Malpezzi, M. Molteni, A. Mele, W. Panzeri, and M. Zanda, Tetrahedron Lett., 50, 2540 (2009).
R. Huisgen, in: A. Padwa (editor), 1,3-Dipolar Cycloaddition Chemistry, Vol. 1., Wiley Interscience, New York (1984), p. 1.
R. Huisgen and G. Mloston, in: A. A. Potekhin, R. R. Kostikov, M. S. Baird (editors), Modern Problems of Organic Chemistry, Vol. 14, St. Petersburg University Press, St. Petersburg (2004), p. 23.
R. Jasiński, M. Kwiatkowska, and A. Barański, Wiad. Chem., 61, 485 (2007).
V. Yu. Korotayev, A. Yu. Barkov, P. A. Slepukhin, M. I. Kodess, and V. Ya. Sosnovskikh, Mendeleev Commun., 21, 112 (2011).
R. Jasiński, Tetrahedron, 69, 927 (2013).
P. Perez, L. R. Domingo, and A. Aizman, and R. Contreras, in: A. Toro-Labbé (editor), Theoretical and Сomputational Chemistry, Vol. 19, Elsevier (2007), p. 139.
P. Perez and E. Chamorro, Lett. Org. Chem., 8, 88 (2011).
P. K. Chattaraj, S. Giri, and S. Duley, Chem. Rev., 111, PR43 (2011).
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Jr. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, Y. Nakajima, O. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, M. C. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian 03, Gaussian Inc., Pittsburgh (2003).
J. Tomasi, B. Mennucci, and E. Cancès, J. Mol. Struct. (Theochem), 464, 211 (1999).
R. Jasiński, O. Koifman, and A. Barański, Mendeleev Commun., 21, 262 (2011).
R. Jasiński and A. Barański, Polish J. Chem., 81, 1441 (2007).
R. Jasiński, K. Wąsik, M. Mikulska, and A. Barański, J. Phys. Org. Chem., 22, 717 (2009).
H. B. Schlegel, J. Comp. Chem., 3, 214 (1982).
V. D. Kiselev and A. I. Konovalov, Usp. Khim., 58, 383 (1989).
V. D. Kiselev and A. I. Konovalov, J. Phys. Org. Chem., 22, 466 (2009).
R. Sustmann, W. Sicking, and R. Huisgen, J. Am. Chem. Soc., 117, 9679 (1995).
G. Leroy, M. Sana, L. A. Burke, and M. T. Nguyen, Quantum Theory Chem. React., 1, 91 (1980).
Author information
Authors and Affiliations
Corresponding author
Additional information
*For communication 16, see [1].
Published in Khimiya Geterotsiklicheskikh Soedinenii, No. 7, pp. 1132-1137, July, 2013.
Rights and permissions
About this article
Cite this article
Jasiński, R., Socha, J. & Barański, A. Conjugated nitroalkenes in cycloaddition reactions. 17*. Mechanism of (2+3) cycloaddition between (E)-3,3,3-trifluoro-1-nitroprop-1-ene and (Z)-N-methyl-C-phenylnitrone in the light of reactivity indices theory and B3LYP/6-31G* calculations. Chem Heterocycl Comp 49, 1055–1060 (2013). https://doi.org/10.1007/s10593-013-1343-9
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-013-1343-9