
Abstract
Invasive species are one of the greatest threats to biodiversity, with endemic species on islands being at particular risk. Management programs can help to minimize these impacts, but such programs are most successful when they are well-informed. In the Galápagos Islands, Ecuador, a recently introduced avian parasitic fly, Philornis downsi, has had strong negative effects on the survival of multiple endemic bird species, including several species of Darwin’s finches. The fly now populates most of the major islands within the Archipelago and the need to better understand the population structure and connectivity patterns of this invasive fly has become increasingly apparent as various management efforts are being considered. Here, we use genomic and phylogenetic approaches to estimate population structure and connectivity for P. downsi collected from five islands within the Galápagos Islands and several sites in mainland Ecuador, which is the presumptive origin of the invasive population. Genomic data showed very little genetic differentiation between island populations of P. downsi relative to the mainland. Phylogenetic analyses, which used more conservative genetic markers than the genomics approach, showed that island and mainland populations of flies were highly related. Our study provides some of the first results using genetic data to quantify differentiation among mainland and island populations of P. downsi. In addition, our study found very little genetic differentiation between island populations of flies, suggesting that there may be considerable gene flow among islands; however, further sampling is needed to determine the extent to which this could be occurring. As management techniques aimed at controlling the impact of this parasite on endemic bird populations are being considered, our study provides important insights into the history of P. downsi’s invasion to the Galápagos Islands and current population connectivity patterns.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Species invasions are occurring more frequently as globalization increases (Hulme 2009; McGeoch et al. 2010). The economic impact of these invasions is estimated to exceed $120 billion annually (Pimentel et al. 2005) and the ecological ramifications of species invasions, which occur at ecosystem, community, and population levels, are perhaps even more staggering (Wilcove et al. 1998; Mooney 2005; Raizada et al. 2008; Vila et al. 2011; Simberloff 2013; Hejda et al. 2017). Invasive species can disrupt ecosystem services (Charles and Dukes 2008; Walsh et al. 2016) and lower biodiversity through predation, competition, parasitism, and disease (Reaser et al. 2007; Rowles and O’Dowd 2007; Vilcinskas et al. 2013). The loss of biodiversity as a consequence of invasive species has been especially well documented on islands, posing a particular threat to endemic species that are unique to these ecosystems (Sax and Gaines 2008; Tershy et al. 2015; Doherty et al. 2016).
Management of target populations of invasive species on islands can alleviate negative effects on native ecosystems (Vreysen et al. 2000; Causton et al. 2005; Cruz et al. 2005; Jones et al. 2016). However, defining target populations based on faulty presumptions of population structure can mislead management efforts and reduce their efficacy. The identification of “eradication units” focuses instead on the eradication or management of genetically isolated clusters of populations (Robertson and Gemmell 2004), such that the risk of reinvasion is minimized. This approach has increased the success of efforts aimed at eradicating rat populations in several insular systems (Abdelkrim et al. 2005; Savidge et al. 2012; Ragionieri et al. 2013). For example, Abdelkrim et al. (2005) used a population genetics approach to show that Norway rats (Rattus norvegicus) are able to regularly migrate between some islands in Archipelagos off the Brittany Coast of France, such that eradication of a “population” on a single island was likely to be highly ineffective. Importantly, this study demonstrated the necessity of understanding population connectivity prior to implementing management strategies.
Philornis downsi is a parasitic nest fly of birds and is invasive to the Galápagos Islands, Ecuador (Kleindorfer and Dudaniec 2016; Fessl et al. 2018; McNew and Clayton 2018). Adult females lay eggs in the nests of 22 of the 28 land bird species that nest there, including 11 of the 17 species of Darwin’s finches (Fessl et al. 2018). The fly larvae are semi-hematophagous parasites of nestlings and adult female birds (Fessl et al. 2006; Cimadom et al. 2016; Common et al. 2019). Nestling mortality can be extremely high but varies by host species and rainfall patterns (Dudaniec et al. 2007; Koop et al. 2013; Cimadom et al. 2014; Heimpel et al. 2017). Philornis downsi has been implicated in the decline of several endemic Galápagos birds, including the Mangrove Finch (Cactospiza heliobates), the Medium Tree-Finch (Camarhynchus pauper), and the Warbler Finch (Certhidia olivacea) (Dvorak et al. 2004; Grant et al. 2005; Cunninghame et al. 2017; Peters and Kleindorfer 2018; Bulgarella et al. 2019). While first recorded in the Galápagos Islands in 1964, P. downsi was not observed in the nests of land birds until 1997 (Fessl and Tebbich 2002). Currently, the fly has been observed on 15 of the 17 major islands sampled within the archipelago (Wiedenfeld et al. 2007; Jimenez-Uzcategui et al. 2011). To date, the source of colonization remains unknown though P. downsi has been documented in Trinidad and Tobago (Dodge and Aitken 1968), Argentina (Silvestri et al. 2010), Brazil (de Carvalho and Couri 1999), and mainland Ecuador (Bulgarella et al. 2015). The Galápagos National Park and the Charles Darwin Foundation have prioritized management efforts for P. downsi in the Galápagos, including exploration of biological control and sterile insect techniques (Bulgarella et al. 2017; Fessl et al. 2018; Boulton et al. 2019). However, these programs can greatly benefit from knowledge of the population genetics of the targeted species.
A major goal of this study is to describe population structure in the native and invasive ranges of this parasite. Very little genetic work has been done on this system, largely due to the inherent difficulties of collecting samples. However, an important study by Dudaniec et al. (2008) used mitochondrial and microsatellite data to assess population structure among three island populations of P. downsi in the Galápagos. This study confirmed that a single species of P. downsi was present on all of the islands tested, as was recently supported by Common et al. (2020) who found no morphological differences between island populations of flies. Importantly, Dudaniec et al. (2008) showed that there were low, but significant levels of genetic differentiation between some of the islands, though clustering analyses did not differentiate between island populations. Furthermore, this study found evidence of a recent genetic bottleneck, consistent with the hypothesis that P. downsi invaded the Galápagos Islands recently, which is also supported by historical considerations (Kleindorfer and Sulloway 2016; Fessl et al. 2018). Our study builds on this work by examining current population structure among island and mainland populations of P. downsi. Here, we use a genomics approach including over 2000 genome-wide SNP markers obtained from restriction-site associated DNA sequencing (RAD-seq) and a phylogenetics approach using two gene sequences to further estimate population structure, phylogenetic history, and possible connectivity patterns for P. downsi collected from the Galápagos Islands and mainland Ecuador.
Methods
A genomics approach: RAD sequence processing and population genetics analyses
Adult P. downsi were collected from five islands within the Galápagos archipelago (Santa Cruz, San Cristobal, Floreana, Isabela, Santiago) between March 2015 and April 2017 (Fig. 1). On Floreana, Isabela, and Santiago, flies were sampled from several locations within each island. For all island sampling, adult flies were caught using McPhail traps (BioQuip Products, California, USA), baited with fermented papaya juice, that were set out for 3–5 days as in Causton et al. (2019). Following collection, flies were stored in 70% ethanol until being transported to the United States for further processing. In addition, P. downsi were collected at a site in mainland Ecuador. This was the Reserva Ecológica Loma Alta in the Province of Santa Elena, Ecuador (hereafter referred to as Loma Alta), where 38 P. downsi pupae were collected from 5 nests between April 5 and 25, 2017; each were identified as P. downsi based upon pupal characteristics (Couri 1999). These pupae were transported to a U.S. government-approved Quarantine Facility at the University of Minnesota under Ecuadorian export and United States import permits, where they were reared to adulthood (as in Bulgarella et al. 2015). These adults were used in laboratory studies prior to being preserved in 70% ethanol. DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen, Valencia, California, USA) on whole fly samples. Samples that returned low yields of DNA (< 10 ng/μl) were not submitted for sequencing and were removed from the study.

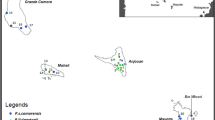
Map of Ecuador, South America and the Galápagos Islands showing sampling locations of P. downsi. For the genomics study (solid circles), samples were collected from a single location from Santa Cruz Island, San Cristobal Island, and Loma Alta, mainland. On three islands, flies were collected from multiple locations and combined for analyses (Floreana Island, 2 locations; Isabela Island, 2 locations; Santiago Island, 2 locations). For the phylogenetics study (open circles), samples were collected from 2 locations on the mainland and 2 locations on Santa Cruz Island. Hashed circles represent locations where samples were collected for both the genomics study and the phylogenetics study
In total, 89 samples were submitted for sequencing. These included P. downsi samples from Santiago (n = 3), Santa Cruz (n = 14), San Cristobal (n = 5), Floreana (n = 8), Isabela (n = 25), and mainland Ecuador, Loma Alta (n = 34) (Table S1). DNA extractions were normalized to 10 ng/μl, loaded individually in 100 μl quantities on a 96-well plate and submitted for sequencing. Single-digest restriction site associated DNA sequencing (RAD-seq) and library preparation were performed by Floragenex (Eugene, Oregon, USA) using the protocol by Etter et al. (2011). Genomic DNA was first digested with the restriction enzyme SbfI (New England Biolabs, Inc., Ipswich, Massachusetts, USA) followed by ligation of a P1 adapter. Multiplexing barcodes were adhered to each sample followed by pooling, shearing and size selection of samples, ligation of a P2 adapter and PCR enrichment. Finally, pooled samples were sequenced on a single lane of a HiSeq 2000 Illumina sequencer (single end 100 bp sequencing).
The software program Stacks v 2.41 was used to process the raw data, call variants, and perform some population-level analyses (Catchen et al. 2013). Raw sequences were demultiplexed, barcodes removed, and low-quality reads were filtered using process_radtags (Catchen et al. 2013). All samples had a sequencing read depth > 20 × (as estimated with the denovo_map.pl program in Stacks); therefore, no samples needed to be removed to avoid misrepresentation of heterozygote diversity (Rochette and Catchen 2017).
There currently is no reference genome for P. downsi; therefore, we used the wrapper script denovo_map.pl within Stacks to execute a pipeline of commands. The wrapper implemented ustacks (builds loci de novo), cstacks (catalogs and matches loci across samples), sstacks (matches samples against the catalog), tsv2bam (transposes data such that it is organized by RAD locus), and gstacks (calls SNPs for each locus) (Catchen et al. 2013). A subset of individuals were analyzed by varying the parameters for maximum number of nucleotide mismatches allowed between stacks within (M) and between (n) individuals (M = n, ranging from 1 to 9) in order to assemble putative alleles and loci, respectively (Paris et al. 2017; Rochette and Catchen 2017). Optimal conditions were determined as the minimum value of M and n that resulted in an asymptotic number of loci being shared by \(\ge\) 80% of the subset individuals. Thus, for final analyses of the full dataset, a maximum of two nucleotide mismatches were allowed between stacks within (M = 2) and between (n = 2) individuals, and the number of reads required to form a stack was set at 3 (M = 3) (Rochette and Catchen 2017).
The populations function in Stacks was used to further filter the data such that the final data set contained loci that were in \(\ge\) 60% of individuals overall and \(\ge\) 80% of individuals within each population and could be found in 3 or more populations. Additionally, the minimum minor allele frequency was set to 0.05 and the maximum observed heterozygosity required to process a SNP was set at 0.70 (Rochette and Catchen 2017). Analyses were restricted to a single random SNP per locus, which can act to reduce linkage disequilibrium. Output files from Stacks were converted using PGDSpider v. 2.1.1.5 (Lischer and Excoffier 2012), then analyzed with BayeScan v.2.1 (Foll and Gaggiotti 2008) to identify loci under selection. Loci with an FDR > 0.1 were removed from further analyses (n = 4 loci). All default options were used, except a prior odds value was set at 100 (default is 10) to avoid an excess of false positives, especially with limited sampling of individuals in some populations (Foll and Gaggiotti 2008). In addition, VCFtools (Danecek et al. 2011) was used to test for SNPs deviating from Hardy–Weinberg Equilibrium in each population. Following all of these filtering steps, 2222 of 328,992 loci were retained for downstream analyses. We explored the data using both less restrictive and more restrictive filtering parameters, none of which drastically altered the main conclusions of our study. Estimates of genetic diversity, including observed heterozygosity (HO) and expected heterozygosity (HE), were calculated with the hierfstat package (Goudet 2005) in R (version 3.4) (R Development Core Team 2013). The number of private alleles per population was calculated with the poppr package in R (Kamvar et al. 2014). Pairwise FST distances [with associated p-values following correction by false discovery rate (FDR)] and measures of variance within and between populations (AMOVA) was calculated using GENODIVE (version 3.0) (Meirmans 2020).
The level of genetic differentiation relative to the geographic distance between populations (i.e. isolation by distance) was tested by comparing FST values to linear geographic distances (km) estimated using Google Earth. Significance was determined using Mantel tests performed in R with the ecodist package (Goslee and Urban 2007), with 10,000 permutations.
The number of genetically distinct clusters needed to describe the data was first estimated using the K-means method generated in STRUCTURE v.2.3.4 (Evanno et al. 2005). Five independent runs (10,000 length burn-in, 100,000 iterations, admixture allowed) were conducted for each K value (K = 1–6). The CLUMPAK online server (https://clumpak.tau.ac.il/contact.html) was used to average iterative runs of K and determine an optimal estimate of the number of distinct genetic clusters using the delta K method (Evanno et al. 2005; Kopelman et al. 2015; Meirmans 2015, 2019; Janes et al. 2017). We complemented this analysis by performing discriminant analysis of principal components (DAPC), which optimizes variance between groups while minimizing variance within groups to identify populations. These analyses were done using the dapc function (Jombart et al. 2010) within the adegenet package in R (Jombart 2008). DAPC performs a discriminant analysis on principal components retained through the cross-validation step implemented using the xvalDapc function.
To address limitations in sample size at three of our island sites, we ran all of these same clustering analyses with a limited data set that included only individuals from Santa Cruz Island (BG), Isabela Island (IS), and the mainland (LA). We hereafter refer to these data sets as the full and limited data sets, respectively. We ran all of the same filtration steps as described above, which resulted in 2193 variable loci being used in the limited dataset, after 29 loci that were non-variant among these 3 populations were removed.
Fastq sequence files have been deposited in the NCBI Short-Read Archive (SRA) database (BioProject Number PRJNA614982; Table S1).
A phylogenetics approach: gene sequencing and phylogenetics analyses
In a separate study, we extracted, amplified, and sequenced 1005 bp of the mitochondrial cytochrome oxidase 1 gene (CO1) and 442 bp of the nuclear ITS2 rDNA gene region (ITS2) for 31 P. downsi flies collected during the 2014/2015 season from two sites in mainland Ecuador (Bosque Protector Cerro Blanco, Guayas Province, n = 11, and Reserva Ecológica Loma Alta, Santa Elena Province, n = 1) and from two sites in Santa Cruz Island, Galápagos (Los Gemelos, n = 14, and Playa El Garrapatero, n = 5) (Fig. 1; Table S1). We used the DNeasy Blood and Tissue Extraction kit (Qiagen, Valencia, California, USA) for DNA extraction and samples were amplified using the forward primer M202 (C1-J-1751, Simon et al. 1994) paired with the reverse primer M70 (UEA10, Lunt et al. 1996) for CO1. For ITS2, we used the primer pair developed for Philornis spp. by Monje et al. (2013). PCR amplifications and Sanger sequencing followed standard protocols (Bulgarella et al. 2015). Sequences from opposite strands were reconciled and verified for accuracy using Sequencher v.5.2.4 (Gene Codes, Ann Arbor, Michigan, USA). We aligned the sequences for each gene using the Geneious v.10.2.3 alignment algorithm (https://www.geneious.com) with the default parameter settings. We concatenated the CO1 and ITS2 data, partitioned by gene region, to obtain a combined dataset of 1529 bp. We conducted maximum likelihood and Bayesian analyses on the combined dataset using a single, best-fit model of sequence evolution as determined by AIC (Akaike 1973) implemented in Modeltest v.3.7 (Posada and Crandall 1998). For maximum likelihood analyses, we completed 1000 full heuristic tree searches in the PAUP*4.0b10 (Swofford 2002) plug-in for Geneious, each with random addition of taxa. Statistical support for nodes was evaluated with nonparametric bootstrapping (Felsenstein 1985) implementing 2000 replicates in PAUP. Trees were rooted with Philornis falsificus as a paraphyletic group from the DNA of a single P. falsificus individual collected at Cerro Blanco in May, 2015 (Bulgarella et al. 2017).
Clade probabilities for the combined 1529 bp dataset were also obtained from the posterior distribution using the MrBayes v.3.2.6 (Huelsenbeck and Ronquist 2001) plug-in for Geneious. Bayesian analyses were replicated twice, each with 4 Markov chains of 2 million generations. Trees were sampled every 2500 generations, of which the first 150,000 generations were discarded as burn-in. Sequences have been archived in GenBank (accession numbers for P. downsi are MT186607–MT186637, MT198952–MT198979, KP730049; for P. falsificus MT229984 and MT240486; Table S1).
Results
Genomics approach
In the full data set (including 6 locations and 2222 variant loci) observed heterozygosity (HO) was similar across all island and mainland populations, ranging from 0.21 to 0.24 (Table S2). Private alleles varied widely across populations and were highest in the mainland population (Loma Alta: 478 private alleles) compared to the islands (private alleles ranged from 1 to 14 per island population) (Table S2). Pairwise genetic distances (FST) between populations ranged from 0.001 to 0.282, and were highest in comparisons between mainland and island populations (Table 1). Analysis of molecular variance comparing all six populations showed that 14.8% of the molecular variance was attributable to variation among populations [FST = 0.148, standard deviation (SD) = 0.005], 14.7% was attributable to variation among individuals (FIS = 0.173, SD = 0.004), and 70.5% was attributable to variation within individuals (FIT = 0.295, SD = 0.005). There was a significant isolation-by-distance effect across all populations (Mantel, r = 0.98, p = 0.02). However, when the mainland population was excluded, there was no longer a significant signature of isolation-by-distance among island samples (Mantel, r = 0.60, p = 0.19).
The K-means method as applied through STRUCTURE indicated an optimal number of K = 2 genetic clusters with the full dataset (Fig. S1). Given documented issues with K = 2 being identified more often than by chance when using the delta K method (Janes et al. 2017), we show the results for K = 2–5 in Fig. 2a. With K = 2, the mainland and island populations differentiate. Then, with increasing values of K, additional genetic variation is found largely within the mainland population cluster. The DAPC method was used to complement analyses using STRUCTURE. In the first step, 100 principal components were retained (Fig. S2a), which accounted for over 98% of the variance. The Bayesian Information Criterion (BIC) was used to identify an optimal number of clusters as K = 2 (Fig. 2b, left). Then, 11 principal components were retained for the DAPC following cross-validation analysis (Fig. S2b) and 1 discriminant function. DAPC shows two main clusters (Fig. 2b, right), and the assignment plot shows that the clusters correlate with mainland versus island individuals (Fig. S2c).

Results of STRUCTURE and DAPC analyses of Philornis downsi sampled from the Galápagos Islands and mainland Ecuador. a Graphical representation of Bayesian clustering approach using STRUCTURE and CLUMPAK to graph a consensus of five iterative runs at K values 2–5, following permutations testing. Each vertical line represents a single individual. Sampling locations are denoted below each plot, representing samples from the Galápagos and mainland. Various colors within the plot represent inferred membership to a given genetic cluster. Posterior probability methods identified K = 2 as the optimal number of distinct genetic clusters within the total sample set. b Plot of the number of clusters in the DAPC analyses using Bayesian Information Criterion (BIC) to select an optimum. The “elbow joint” in the plot indicates the optimal number of clusters to select as it represents the lowest BIC value. c DAPC scatter plot of 2 main clusters based on 11 retained PCs and 1 discriminant function. As with the STRUCTURE analysis, the distinction between these clusters is based on geographic origin of the samples (i.e. mainland or islands)
To address low sample size for 3 island populations, we performed these same clustering analyses on a limited dataset that included only locations with a sample size \(\ge\) 14 individuals [i.e. the islands of Santa Cruz, Isabela, and mainland Ecuador (Loma Alta)] with 2193 variant loci. The K-means method as applied through STRUCTURE again indicated an optimal number of K = 2 genetic clusters (Figs. S3, S4a). At K = 2, there is clear differentiation between samples from the Galápagos Islands and those from the mainland. Increasing values of K show increasing genetic variation largely within the mainland population. Eighty PCs were retained in the first step of the DAPC method (Fig. S4b, top left). The BIC was used to identify an optimal number of clusters as K = 2 (Fig. S4b top right). Following cross-validation (Fig. S4b, bottom left), 10 PCs were retained and 1 discriminant function, which resulted in 2 clusters (Fig. S4b, bottom right).
Phylogenetics approach
For the combined dataset, the best fit model of nucleotide substitution was a transition model (TIM) with variable base frequencies, variable transition rates and two transversion rates. The percent of identical sites for the 31 P. downsi flies sampled from all sites was 99.6% showing little genetic variability. Maximum likelihood analyses showed that P. downsi collected in mainland Ecuador and the Galápagos Islands form a monophyletic group with 100% bootstrap support (Fig. 3). We found the same result when we performed a Bayesian analysis. The monophyly of P. downsi was supported by a posterior probability of 1 (not shown).

Maximum likelihood tree of 31 P. downsi specimens from mainland Ecuador (Guayaquil and Loma Alta), the Galápagos Islands (Santa Cruz Island), rooted with the closely related species, Philornis falsificus collected in Guayaquil, Ecuador. Support values above branches correspond to nonparametric bootstrapping
Discussion
We used genomic and phylogenetic approaches to assess the population structure of P. downsi sampled from its native and invasive ranges. Our primary goal was to better understand patterns of genetic differentiation between mainland and island populations of flies and between island populations of flies. We consistently found genetic differentiation between mainland and island populations using the genomics approach. Phylogenetic comparisons using CO1 and ITS2 genes showed almost no differentiation between mainland and island populations of flies, though this is likely due in part to the more conserved nature of these genetic markers. In addition, we found very little genetic differentiation between island populations of flies and no pattern of isolation by distance across island populations. Together, our results suggest that population connectivity between mainland and island populations is likely limited, but there is the potential for continued gene flow among island populations that warrants further investigation. It is important to note that in regard to the lack of differentiation found between island populations, we did not explicitly test whether this pattern is the consequence of a recent invasion or ongoing migration of flies between islands. Thus, we will discuss the consequences of either or both being true.
Multiple lines of evidence support the hypothesis that P. downsi invaded the Galápagos Islands relatively recently. The first report of this species in the Galápagos dates from 1964 (Kleindorfer and Dudaniec 2016; Fessl et al. 2018) and multiple prior insect surveys failed to report this species (Linsley and Usinger 1966; Fessl et al. 2018). The first observation of P. downsi in a finch nest was made in 1997 (Fessl et al. 2001), despite inspections of nests prior to this (Lack 1947; Grant 1999). In addition, Kleindorfer and Sulloway (2016) inspected museum specimens of Small Ground-Finches (Geospiza fuliginosa) from Floreana Island and only found evidence of beak scarring after 1960. First-instar larvae of P. downsi can feed in the naris of nestlings, leading to scarring and malformations that persist into adulthood (Kleindorfer and Sulloway 2016). Finally, Dudaniec et al. (2008) found minimal genetic differentiation between island populations and an excess of heterozygosity across all individuals using eight microsatellites markers, a pattern consistent with a very recent genetic bottleneck.
Our study found further evidence supporting the hypothesis that P. downsi recently invaded the Galápagos. We found that mainland flies clustered as a separate population from island flies across multiple genomic analyses. The mainland fly population had significantly higher levels of genetic diversity (e.g. number of private alleles) than did the Galápagos populations, which is expected if mainland Ecuador represents the native range. Phylogenetic comparisons showed almost no differentiation between mainland and island populations of flies, and this result is expected under the premise of a recent invasion, given the conserved nature of these genetic markers. Here, we chose sites on the mainland for their close proximity to the city of Guayaquil, Ecuador. Guayaquil is and has served as the port city for most of the air and boat traffic to the Galápagos, which is a suspected mechanism of introduction for P. downsi (Bulgarella et al. 2015, 2017; Fessl et al. 2018). Further sampling of mainland populations of P. downsi could alter the patterns of diversification shown by our study, though they are unlikely to refute the recent invasion hypothesis. That is to say, additional sampling on the mainland may reveal a population that is more genetically similar to island populations than what we have sampled here. Significant further study is needed to explore potential mainland source populations and resolve a complete invasion history of this parasite.
Low levels of genetic differentiation can be the result of a recent invasion in which genetic drift or selective sweeps have had minimal time to fix alleles within populations. Similarly, we expect to find low levels of differentiation when gene flow is persistent among populations. While our study was not designed to differentiate between these two non-mutually exclusive hypotheses, our results suggest that ongoing gene flow between island populations may be occurring. Most of the genetic variation observed in our study was explained at the level of individuals, consistent with analyses showing relatively low pairwise genetic differentiation between island populations. Under conditions of limited gene flow, a pattern of isolation by distance is expected to occur relatively quickly, yet we did not find evidence of this pattern among island populations. Our results, particularly those using the limited dataset, are largely consistent with Dudaniec et al. (2008), who used eight microsatellite markers in a landscape genetic analysis to show limited, but significant genetic differentiation (FST values) between Floreana Island and Santa Cruz Island, Floreana Island and Isabela Island, but not between Santa Cruz Island and Isabela Island. Similarly, cluster analyses of this same dataset did not detect structure among the three island populations (Dudaniec et al. 2008). It is important to note that sampling from several of our island populations was limited, making our conclusions about population connectivity between these other islands more speculative. A more detailed study on Floreana Island showed varying levels of genetic relatedness among flies based on habitat type (Dudaniec et al. 2010), demonstrating that additional sampling has the potential to reveal more fine scale genetic structure than was shown by our full or limited datasets. Thus, additional sampling within and among islands will facilitate more accurate estimates of gene flow within the archipelago and is highly recommended.
The mechanism by which flies may be dispersing between the islands remains unknown. Adult flies are strong fliers and may be able to move between islands on air currents. Transport via air and boat vessels is also quite likely. Philornis downsi has been documented on boats moving between islands (Lomas 2008), though very infrequently. Boat and air traffic between islands and between islands and the mainland are frequent and increasing (Toral-Granda et al. 2017). Four of the five islands we sampled are inhabited with permanent human residents (Santa Cruz, San Cristobal, Isabela, Floreana) and the fifth island (Santiago) is a popular tourist destination with multiple boats arriving daily for day-hikes. Therefore, ample opportunities exist for interisland transport of flies via boats. The introduction of P. downsi from the mainland to the islands also presumably occurred via boats or airplanes, although further details remain unknown. Biosecurity measures aimed at reducing alien species introductions to the Galápagos have been put into place relatively recently, including spraying pesticides on arriving planes and boats. While the Galápagos now relies on its own biosecurity agency, considerable investment is still needed to have a significant impact on reducing the risks of new or repeated introductions (Toral-Granda et al. 2017).
Management implications
Philornis downsi presents an imminent threat to the endemic Galápagos avifauna with modelling studies suggesting the possibility of extinction of even the more common bird species within a century (Koop et al. 2015). Treatments with insecticides, either through direct application to nests or through inoculated cotton that the birds place in nests themselves, are approaches that have been demonstrated to effectively reduce parasite abundance and improve fledging success (Koop et al. 2011; Knutie et al. 2014). However, these approaches are only a short-term solution for protecting birds in small areas and may introduce risks to birds and other non-target organisms (Causton et al. 2019). Thus, long-term integrated management techniques are under development, and consideration of aspects of P. downsi population genetics and phylogeography could greatly aid these efforts.
One such management technique involves the potential introduction of highly-specialized parasitoid wasps to control P. downsi (Bulgarella et al. 2017; Heimpel 2017; Boulton et al. 2019). Having an understanding of the location within the native range from which the Galápagos populations originated can inform such efforts by directing parasitoid collecting to appropriate mainland locations and gaining information on the coevolution of P. downsi and its parasitoids (Hufbauer and Roderick 2005; Heimpel and Mills 2017). A sterile insect program is also being considered, which involves the field-release of many lab-reared sterilized male flies with the aim that they breed with wild females, who then fail to produce offspring. These programs have been highly successful in the control of a number of other invasive fly species (Hendrichs et al. 1995; Vreysen et al. 2000; Klassen and Curtis 2005). For the sterile insect technique, an understanding of P. downsi population genetics would be crucial since the method is very sensitive to factors such as repeated introductions and multiple mating (Krafsur 2005). Finally, the development of attractants for use in fly traps is also underway (Cha et al. 2016; Mieles 2018) and an understanding of dispersal patterns—such as the possibility of migration from highland to lowland sites on a single island—would greatly enhance the interpretation of trap catch data (Causton et al. 2019).
The intention of our study was to provide insight on the population structure and connectivity of P. downsi in the Galápagos Islands and mainland Ecuador. Our results suggest that with respect to managing P. downsi, the archipelago may need to be treated as a single management unit (Robertson and Gemmell 2004), as several studies have now found little evidence of isolation among island populations of flies (Dudaniec et al. 2008). Efforts to eliminate or manage populations, irrespective of the specific technique, would need to involve simultaneous treatment of islands since efforts to control populations on a single island could be thwarted by recolonization from a neighboring island. The use of modern genetic resources to indirectly assess patterns of gene flow between populations has great potential to improve management efforts across biological systems. In the Galápagos, further sampling will allow for more detailed estimates of gene flow between the islands and perhaps, even within individual islands (Dudaniec et al. 2010). Furthermore, these same genetic tools can be used to identify the source population(s) and the most likely mechanism of invasion, allowing managers to reduce the likelihood of reinvasion from the mainland.
References
Abdelkrim J, Pascal M, Calmet C, Samadi S (2005) Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations. Conserv Biol 19:1509–1518
Akaike H (1973) Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F (eds) Proceedings of second international symposium on information theory. Akademia Kiado, Budapest, pp 267–281
Boulton RA, Bulgarella M, Ramirez IE, Causton CE (2019) Management of the invasive avian parasitic fly, Philornis downsi, in the Galapagos Islands: is biological control a viable option? In: Veitch CR, Clout MN, Martin AR, Russell JC, West CJ (eds) Island Invasives: scaling up to meet the challenge. IUCN, Gland, pp 360–363
Bulgarella M, Quiroga MA, Boulton RA, Ramirez IE, Moon RD, Causton CE, Heimpel GE (2017) Life cycle and host specificity of the parasitoid Conura annulifera (Hymenoptera: Chalcididae), a potential biological control agent of Philornis downsi (Diptera: Muscidae) in the Galapagos Islands. Ann Entomol Soc Am 110:317–328
Bulgarella M, Quiroga MA, Brito vera GA, Dregni JS, Cunningham F, Mosquera Munoz DA, Monje LD, Causton CE, Heimpel GE (2015) Philornis downsi (Diptera: Muscidae), an avian nest parasite invasive to the Galapagos Islands, in mainland Ecuador. Ann Entomol Soc Am 108(3):248–250
Bulgarella M, Quiroga MA, Heimpel GE (2019) Additive negative effects of Philornis nest parasitism on small and declining Neotropical bird populations. Bird Conserv Int 29:339–360
Catchen J, Hohenlohe P, Bassham S, Amores A, Cresko W (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Causton CE, Moon RD, Cimadom A, Boulton RA, Cedeno D, Lincango MP, Tebbich S, Ulloa A (2019) Population dynamics of an invasive bird parasite, Philornis downsi (Diptera: Muscidae), in the Galapagos Islands. PLoS ONE 14:e0224125
Causton CE, Sevilla CR, Porter SD (2005) Eradication of the little fire ant, Wasmannia auropunctata (Hymenoptera: Formicidae), from Marchena Island, Galapagos: on the edge of success? Fla Entomol 88:159–168
Cha DH, Mieles AE, Lahuatte PF, Cahuana A, Lincango MP, Causton CE, Tebbich S, Cimadom A, Teale SA (2016) Identification and optimization of microbial attractants for Philornis downsi, an invasive fly parasitic on Galapagos birds. J Chem Ecol 42:1101–1111
Charles H, Dukes JS (2008) Impacts of invasive species on ecosystem services. Springer, Berlin
Cimadom A, Causton C, Cha DH, Damiens D, Fessl B, Hood-Nowotny R, Lincango P, Mieles AE, Nemeth E, Semler EM, Teale SA, Tebbich S (2016) Darwin’s finches treat their feathers with a natural repellent. Sci Rep 6:34559
Cimadom A, Ulloa A, Meidl P, Zottl M, Zottl E, Fessl B, Nemeth E, Dvorak M, Cunninghame F, Tebbich S (2014) Invasive parasites, habitat change and heavy rainfall reduce breeding success in Darwin’s finches. PLoS ONE 9(9):e107518
Common LK, Dudaniec RY, Colombelli-Negrel D, Kleindorfer S (2019) Taxonomic shifts in Philornis larval behaviour and rapid changes in Philornis downsi Dodge & Aitken (Diptera: Muscidae): an invasive avian parasite on the Galápagos Islands. In: Sarwar M (ed) Life cycle and development of Diptera. InTech Open, London
Common LK, O’Connor JA, Dudaniec RY, Peters KJ, Kleindorfer S (2020) Evidence for rapid downward fecundity selection in an ectoparasite (Philornis downsi) with earlier host mortality in Darwin’s finches. J Evol Biol 33(4):524–533
Couri MS (1999) Myiasis caused by obligatory parasites. 1a. Philornis Meinert (Muscidae). In: Guimaraes JH, Papavero N (eds) Myiasis in man and animals in the Neotropical region. Bibliographic data base. FAPESP, Sao Paulo, pp 51–70
Cruz F, Donlan CJ, Campbell K, Carrion V (2005) Conservation action in the Galapagos: feral pig (Sus scrofa) eradication from Santiago Island. Biol Conserv 121:473–478
Cunninghame F, Fessl B, Sevilla CR, Young GR, La Greco N (2017) Manejo de la conservacion a largo plazo para salvar al pinzon de manglar (Camarhynchus heliobates) en peligro critico de extincion. In: Informe Galapagos 2015–2016. DPNG, CGREG, FCD and GC, Puerto Ayora, Galapagos, pp 163–170
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, Genomes Project Analysis Group (2011) The variant call format and VCF tools. Bioinformatics 27:2156–2158
de Carvalho M, Couri M (1999) New associations between Philornis Meinert (Diptera, Muscidae) and Thamnophilidae (Aves, Passeriformes). Rev Bras Zool 16(4):116
Dodge HR, Aitken THG (1968) Philornis flies from Trinidad (Diptera: Muscidae). J Kans Entomol Soc 41:134–154
Doherty TS, Glen AS, Nimmo DG, Ritchie EG, Dickman CR (2016) Invasive predators and global biodiversity loss. Proc Natl Acad Sci USA 113:11261–11265
Dudaniec RY, Fessl B, Kleindorfer S (2007) Interannual and interspecific variation in intensity of the parasitic fly, Philornis downsi, in Darwin’s finches. Biol Conserv 139:325–332
Dudaniec RY, Gardner MG, Donnellan S, Kleindorfer S (2008) Genetic variation in the invasive avian parasite, Philornis downsi (Diptera, Muscidae) on the Galapagos Archipelago. BMC Ecol 8:13
Dudaniec RY, Gardner MG, Kleindorfer S (2010) Offspring genetic structure reveals mating and nest infestation behaviour of an invasive parasitic fly (Philornis downsi) of Galapagos birds. Biol Invasions 12:581–592
Dvorak M, Vargas H, Fessl B, Tebbich S (2004) On the verge of extinction: a survey of the mangrove finch Cactospiza heliobates and its habitat on the Galápagos Islands. Oryx 38:1–9
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA (2011) SNP discovery and genotyping for evolutionary genetics using RAD sequencing. In: Orgogozo V, Rockman MV (eds) Molecular methods for evolutionary genetics SE-9. Humana Press, Totowa, pp 157–178
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Felsenstein J (1985) Confidence-limits on phylogenies—an approach using the bootstrap. Evolution 39:783–791
Fessl B, Couri MS, Tebbich S (2001) Philornis downsi Dodge & Aitken, new to the Galapagos Islands, (Diptera, Muscidae). Stud Dipterol 8:317–322
Fessl B, Heimpel G, Causton C (2018) Invasion of an avian nest parasite, Philornis downsi, to the Galapagos Islands: colonization history, adaptations to novel ecosystems, and conservation challenges. In: Parker P (ed) Disease ecology, social and ecological interactions in the Galapagos Islands. Springer, Cham
Fessl B, Sinclair BJ, Kleindorfer S (2006) The life-cycle of Philornis downsi (Diptera: Muscidae) parasitizing Darwin’s finches and its impacts on nestling survival. Parasitology 133:739–747
Fessl B, Tebbich S (2002) Philornis downsi—a recently discovered parasite on the Galápagos Archipelago—a threat for Darwin’s finches? Ibis 144:445–451
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Softw 22:1–19
Goudet J (2005) hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186
Grant PR (1999) Ecology and evolution of Darwin’s finches. Princeton University Press, Princeton
Grant PR, Grant BR, Petren K, Keller LF (2005) Extinction behind our backs: the possible fate of one of the Darwin’s finch species on Isla Floreana, Galápagos. Biol Conserv 122:499–503
Heimpel GE (2017) Could biological control protect Darwin’s finches from an invasive parasite? Biocontrol News Inf 38:21N–22N
Heimpel GE, Hillstrom A, Freund D, Knutie SA, Clayton DH (2017) Invasive parasites and the fate of Darwin’s finches in the Galapagos Islands: the case of the vegetarian finch (Platyspiza crassirostris). Wilson J Ornithol 129:345–349
Heimpel GE, Mills NJ (2017) Biological control: ecology and applications. Cambridge University Press, Cambridge
Hejda M, Hanzelka J, Kadlec T, Strobl M, Pysek P, Reif J (2017) Impacts of an invasive tree across trophic levels: species richness, community composition and resident species’ traits. Divers Distrib 23:997–1007
Hendrichs HJ, Franz G, Rendon P (1995) Increased effectiveness and applicability of the sterile insect technique through male-only releases for control of Mediterranean fruit flies during fruiting seasons. J Appl Entomol 119:371–377
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Hufbauer RA, Roderick GK (2005) Microevolution in biological control: mechanisms, patterns, and processes. Biol Control 35:227–239
Hulme PE (2009) Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46:10–18
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602
Jimenez-Uzcategui G, Llerena W, Milstead WB, Lomas EE, Wiedenfeld DA (2011) Is the population of Floreana Mockingbird Mimus trifasciatus declining? Cotinga 33(1):1–7
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jones HP, Holmes ND, Butchart SHM, Tershy BR, Kappes PJ, Corkery I, Aguirre-Munoz A, Armstrong DP, Bonnaud E, Burbidge AA, Campbell K, Courchamp F, Cowan PE, Cuthbert RJ, Ebbert S, Genovesi P, Howald GR, Keitt BS, Kress SW, Miskelly CM, Oppel S, Poncet S, Rauzon MJ, Rocamora G, Russell JC, Samaniego-Herrera A, Seddon PJ, Spatz DR, Towns DR, Croll DA (2016) Invasive mammal eradication on islands results in substantial conservation gains. Proc Natl Acad Sci USA 113:4033–4038
Kamvar ZN, Tabima JF, Grunwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281
Klassen W, Curtis C (2005) History of the sterile insect technique. In: Dyck V, Hendrichs J, Robinson A (eds) Sterile insect technique principles and practice in area-wide integrated pest management. Springer, Dordrecht, pp 3–36
Kleindorfer S, Dudaniec RY (2016) Host–parasite ecology, behavior and genetics: a review of the introduced fly parasite Philornis downsi and its Darwin’s finch hosts. BMC Zool 1:1
Kleindorfer S, Sulloway F (2016) Naris deformation in Darwin’s finches: experimental and historical evidence for a post-1960s arrival of the parasite Philornis downsi. Glob Ecol Conserv 7:122–131
Knutie SA, McNew SM, Bartlow AW, Vargas DA, Clayton DH (2014) Darwin’s finches combat introduced nest parasites with fumigated cotton. Curr Biol 24:R355–R356
Koop JAH, Huber SK, Laverty SM, Clayton DH (2011) Experimental demonstration of the fitness consequences of an introduced parasite of Darwin’s finches. PLoS ONE 6:e19706. https://doi.org/10.11371/journal.pone.0019706
Koop JAH, Kim PS, Knutie SA, Adler F, Clayton DH (2015) Introduced parasitic fly may lead to local extinction of Darwin’s finch populations. J Appl Ecol 53(2):511–518
Koop JAH, Le Bohec C, Clayton DH (2013) Dry year does not reduce invasive parasitic fly prevalence or abundance in Darwin’s finch nests. Rep Parasitol 3:11–17
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191
Krafsur ES (2005) Role of population genetics in the sterile insect technique. In: Dyck VA, Hendrichs J, Robinson AS (eds) Sterile insect technique: principles and practice in area-wide integrated pest management. Springer, Dordrecht, pp 389–406
Lack D (1947) Darwin’s finches: an essay on the general biological theory of evolution. Cambridge University Press, Cambridge
Linsley E, Usinger R (1966) Insects of the Galapagos Islands. In: Proceedings of the California Academy of Sciences
Lischer HE, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Lomas EE (2008) Dispersion de insectos por las luces de los barcos en las islas Galapagos: una prioridad de conservacion. Universidad central del Ecuador y Fundacion Charles Darwin
Lunt DH, Zhang DX, Szymura JM, Hewitt GM (1996) The insect cytochrome oxidase I gene: evolutionary patterns and conserved primers for phylogenetic studies. Insect Mol Biol 5:153–165
McGeoch MA, Butchart SHM, Spear D, Marais E, Kleynhans EJ, Symes A, Chanson J, Hoffmann M (2010) Global indicators of biological invasion: species numbers, biodiversity impact and policy responses. Divers Distrib 16:95–108
McNew SM, Clayton DH (2018) Alien invasion: biology of Philornis flies highlighting Philornis downsi, an introduced parasite of Galapagos birds. Annu Rev Entomol 63:369–387
Meirmans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24:3223–3231
Meirmans PG (2019) Subsampling reveals that unbalanced sampling affects STRUCTURE results in a multi-species dataset. Heredity (Edinb) 122:276–287
Meirmans PG (2020) genodive version 3.0: easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol Ecol Resour 20(4):1126–1131
Mieles AE (2018) Semiochemical attractants of the parasitic fly Philornis downsi in the Galapagos Islands. State University of New York
Monje LD, Quiroga M, Manzoli D, Couri MS, Silvestri L, Venzal JM, Cuervo P, Beldomenico PM (2013) Sequence analysis of the internal transcribed spacer 2 (ITS2) from Philornis seguyi (Garcia, 1952) and Philornis torquans (Nielsen, 1913) (Diptera: Muscidae). Syst Parasitol 86:43–51
Mooney HA (2005) Invasive Alien Species: the nature of the problem. In: Mooney HA, Mack RN, McNeely JA, Neville LE, Schei PJ, Waage JK (eds) Invasive alien species: a new synthesis. Island Press, Washington, DC, pp 1–15
Paris JR, Stevens JR, Catchen JM (2017) Lost in parameter space: a road map for stacks. Methods Ecol Evol 8:1360–1373
Peters KJ, Kleindorfer S (2018) Avian population trends in Scalesia forest on Floreana Island (2004–2013): Acoustical surveys cannot detect hybrids of Darwin’s tree finches Camarhynchus spp. Bird Conserv Int 28:319–335
Pimentel D, Zuniga R, Morrison D (2005) Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol Econ 52:273–288
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
R Development Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Ragionieri L, Cutuli G, Sposimo P, Spano G, Navone A, Capizzi D, Baccetti N, Vannini M, Fratini S (2013) Establishing the eradication unit of Molara Island: a case of study from Sardinia, Italy. Biol Invasions 15:2731–2742
Raizada P, Raghubanshi AS, Singh JS (2008) Impact of invasive alien plant species on soil processes: a review. Proc Natl Acad Sci India B 78:288–298
Reaser JK, Meyerson LA, Cronk Q, De Poorter M, Eldrege LG, Green E, Kairo M, Latasi P, Mack RN, Mauremootoo J, O’Dowd D, Orapa W, Sastroutomo S, Saunders A, Shine C, Thrainsson S, Vaiutu L (2007) Ecological and socioeconomic impacts of invasive alien species in island ecosystems. Environ Conserv 34:98–111
Robertson BC, Gemmell NJ (2004) Defining eradication units to control invasive pests. J Appl Ecol 41:1042–1048
Rochette NC, Catchen JM (2017) Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc 12:2640–2659
Rowles AD, O’Dowd DJ (2007) Interference competition by Argentine ants displaces native ants: implications for biotic resistance to invasion. Biol Invasions 9:73–85
Savidge JA, Hopken MW, Witmer GW, Jojola SM, Pierce JJ, Burke PW, Piaggio AJ (2012) Genetic evaluation of an attempted Rattus rattus eradication on Congo Cay, U.S. Virgin Islands, identifies importance of eradication units. Biol Invasions 14:2343–2354
Sax DF, Gaines SD (2008) Species invasions and extinction: the future of native biodiversity on islands. Proc Natl Acad Sci USA 105:11490–11497
Silvestri L, Antoniazzi L, Couri MS, Monje L, Beldomenico P (2010) First record of the avian ectoparasite Philornis downsi Dodge and Aitken, 1968 (Diptera: Muscidae) in Argentina. Syst Parasitol 80:137–140
Simberloff D (2013) Invasive species: what everyone needs to know. Oxford University Press, New York
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene-sequences and a compilation of conserved polymerase chain-reaction primers. Ann Entomol Soc Am 87:651–701
Swofford DL (2002) PAUP*: Phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland
Tershy BR, Shen KW, Newton KM, Holmes ND, Croll DA (2015) The importance of islands for the protection of biological and linguistic diversity. Bioscience 65:592–597
Toral-Granda MV, Causton CE, Jager H, Trueman M, Izurieta JC, Araujo E, Cruz M, Zander KK, Izurieta A, Garnett ST (2017) Alien species pathways to the Galapagos Islands, Ecuador. PLoS ONE 12:e0184379
Vila M, Espinar JL, Hejda M, Hulme PE, Jarosik V, Maron JL, Pergl J, Schaffner U, Sun Y, Pysek P (2011) Ecological impacts of invasive alien plants: a meta-analysis of their effects on species, communities and ecosystems. Ecol Lett 14:702–708
Vilcinskas A, Stoecker K, Schmidtberg H, Rohrich CR, Vogel H (2013) Invasive Harlequin ladybird carries biological weapons against native competitors. Science 340:862–863
Vreysen MJB, Saleh KM, Ali MY, Abdulla AM, Zhu ZR, Juma KG, Dyck VA, Msangi AR, Mkonyi PA, Feldmann HU (2000) Glossina austeni (Diptera: Glossinidae) eradicated on the Island of Unguja, Zanzibar, using the sterile insect technique. J Econ Entomol 93:123–135
Walsh JR, Carpenter SR, Vander Zanden MJ (2016) Invasive species triggers a massive loss of ecosystem services through a trophic cascade. Proc Natl Acad Sci USA 113:4081–4085
Wiedenfeld DA, Jimenez GA, Fessl B, Kleindorfer S, Valarezo JC (2007) Distribution of the introduced parasitic fly Philornis downsi (Diptera, Muscidae) in the Galapagos Islands. Pac Conserv Biol 13:14–19
Wilcove DS, Rothstein D, Dubow J, Phillips A, Losos E (1998) Quantifying threats to imperiled species in the United States. Bioscience 48:607–615
Acknowledgements
We thank Gabriel Brito, Sarah Knutie, David Anchundia, Francesca Cunninghame, Birgit Fessl, Paola Lahuatte, Courtney Pike and Ismael Ramirez for collecting and processing the flies. Thanks to Genevieve Kozak and Christy Wails for their assistance with data analyses. Permission to conduct this study was granted by the Galápagos National Park Directorate (Project PC-35-19, PC-07-18 and PC-08-17: Control of the Invasive Parasite, P. downsi and its Impact on Biodiversity) and the Ecuadorian Ministry of the Environment (MAE-DNB-CM-2016-0043). This work was funded by a Grant from the Galápagos Conservancy (Award Number 1-68-308), International Community Foundation (with a Grant awarded by The Leona M. and Harry B. Helmsley Charitable Trust) (Award Number 20140045), Lindblad Expeditions-National Geographic (Award Number 1-01-106) and National Geographic Research and Exploration Grant (Award Number 9847-16). This is contribution number 2348 of the Charles Darwin Foundation for the Galápagos Islands.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10592_2020_1315_MOESM2_ESM.xlsx
Supplementary file2 (XLSX 9 kb) Table S1 Locality and specimen information for the Philornis downsi samples included in thegenomics and phylogenetic studies
Rights and permissions
About this article

Cite this article
Koop, J.A.H., Causton, C.E., Bulgarella, M. et al. Population structure of a nest parasite of Darwin’s finches within its native and invasive ranges. Conserv Genet 22, 11–22 (2021). https://doi.org/10.1007/s10592-020-01315-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01315-0