
Abstract
Understanding patterns of genetic diversity at the landscape scale will enhance conservation and management of natural populations. Here we analyzed the genetic diversity, population connectivity, and spatial genetic structure among subpopulations and age groups of Olea europaea subsp. cuspidata, a cornerstone species of the Afromontane highlands. The study was conducted at the landscape level within a radius of approximately 4 km, as well as on a fine scale (intensive study plot) of less than 300 m radius. In total 542 samples from four natural subpopulations in northwestern Ethiopia were analyzed using ten nuclear microsatellite markers. Inbreeding was higher in smaller populations. No genetic difference was detected among cohorts of different tree sizes in the intensive studied plot. Average population differentiation was low but significant (F ST = 0.016). Landscape genetic analysis inferred two groups: the most distant subpopulation WE located less than 4 kms from the other three subpopulations formed a separate group. Sixty-four percent of the total migrants were shared among the three latter subpopulations, which are spatially clustered. Immigrants were non-randomly distributed inside of the intensive study plot. Significant spatial genetic structure (SGS) was found both at the landscape scale and in the intensive study plot, and adults showed stronger SGS than young trees. An indirect estimate of 220 m as mean gene dispersal distance was obtained. We conclude that even under fragmentation migration is not disrupted in wild olive trees and that large protected populations at church forests are very important to conserve genetic resources. However, the higher level of inbreeding and evidence for population bottlenecks in the small populations, as well as the persisting heavy pressure on most remaining populations, warrants quick action to maintain genetic diversity of wild olive in the Ethiopian highlands.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Even small populations can play a significant role in maintaining genetic diversity of a plant species in a fragmented landscape (Young et al. 1996; Wang et al. 2011). The dynamics of populations in such circumstances have been best described as metapopulation dynamics (Hanski 1998). The influence of natural and artificial barriers on gene flow between population fragments has led to the development of a specific field of research, i.e. landscape genetics (e.g. Holderegger et al. 2010). The long-term fate of fragmented populations strongly depends on habitat availability and ability to migrate among habitat patches. Migration will maintain genetic diversity in a given species through counteracting the negative effect of bottlenecks, genetic drift and elevated levels of inbreeding (Young et al. 1996). Therefore, knowledge about the genetic diversity and connectivity is of high value for conservation and management of disjunct subpopulations (Mills and Allendorf 1996; Keller et al. 2015).
Seed dispersal is an important component of genetic connectivity among neighbouring subpopulations. However, at the landscape level still only few studies are available on the extent and pattern of seed migration. In depth spatial and temporal analysis of migration patterns provides essential evidence about the ecology of seed dispersal. Seed migration is influenced by factors such as availability of habitat, edge effects and isolation by distance (Nathan et al. 2008). In animal dispersed seeds, migration can also be affected by behaviour and habitat preferences of seed dispersers (Jordano et al. 2007).
Limited migration among population fragments eventually will result in the formation of genetic structure (Balkenhol et al. 2016). Classical population genetic studies use statistics like F ST to estimate the amount of differentiation among subpopulations. Current landscape genetic methods widely implement Bayesian approaches using the spatial position of individuals to analyse the genetic structure which allows the detection of landscape barriers to gene flow (Anderson et al. 2010).
In natural populations it is often the most prudent way to assess the dispersal ability and gene flow “indirectly” through the analysis of genetic structure at different spatial scales (Vekemans and Hardy 2004; Hardy et al. 2006). The amount and pattern of spatial genetic structure (SGS) is assessed through computation of pairwise relatedness coefficients between individuals and regressing them on the distance among them (Vekemans and Hardy 2004). Spatial genetic structure in natural populations is a function of multiple forces: major factors such as gene dispersal, mating system and life form (Oddou-Muratorio et al. 2004; Vekemans and Hardy 2004), and other complex reasons such as local seed shadow overlap (Fuchs and Hamrick 2010) and microhabitat heterogeneity (Cruse-Sanders and Hamrick 2004) may significantly influence the formation of SGS. The level of genetic structure and diversity may also vary among generations (Cruse-Sanders and Hamrick 2004; Collevatti et al. 2014). Understanding these temporal patterns is highly relevant for appropriate inference of effects of landscape change on fragmented populations.
The Ethiopian highlands were covered by Juniperus and Olea dominated forest vegetation until the fourteenth to seventeenth century. Later, due to intensive deforestation and soil erosion during the last three centuries the habitat has been changed into a degraded landscape (Darbyshire et al. 2003). Currently, natural forests in the fragmented highlands are usually found only around churches, where cutting of trees is forbidden (Wassie et al. 2010). The impact of humans on these remnant forests continues due to the very high wood demand in the region. In northern Ethiopia only approximately 14% of the total wood demand is supplied domestically (Desalegn and Tadesse 2010). Planting of exotic trees species in Ethiopia is controversially discussed due to ecological disadvantages; therefore, native tree species such as wild olive are needed not only to restore the ecological stability of Ethiopian highlands, but also to provide wood supply and income to rural people.
Wild olive (Olea europaea L. subsp. cuspidata [Wall. ex DC.] Ciferi) is one of six subspecies of the Olea europaea complex. This tree species is native and widely distributed in southern to north-eastern Africa, but also in Southwest Asia, and the drier regions of Yunnan and Sichuan in China (Green 2002). It is a diploid (2n = 46) (Besnard et al. 2008), andromonoecious and mainly wind pollinated species (Breton and Bervillé 2013). The main seed dispersers are birds and small mammals (Cuneo and Leishman 2006; Abiyu et al. 2015). Wild olive is one of the characteristic tree species of the dry Afromontane vegetation type in Ethiopia (Bekele 2005). The occurrence of Olea in Ethiopia is under high pressure due to continuous deforestation of the dry Afromontane vegetation over centuries for agricultural land and fuel wood, and high demand of the wood for farm tool preparation, timber, and in particular charcoal. The species is found in patches of fragmented populations of different size and protection status around old churches and monasteries which are distributed all over the highlands of northern Ethiopia (Aerts et al. 2016). It is one of the main tree species in natural forests (Wassie et al. 2010). Wild olive is also one of the key native tree species for environmental rehabilitation programs, because it can successfully regenerate even on bare lands (Bekele 2005). The remnant church (monastery) forests in the region are found within an average radius of approximately two kilometers to each other (Aerts et al. 2016). Understanding the key microevolationary processes such as gene flow among these forest patches will have a broader regional relevance for management and conservation of forest fragments and for reforestation efforts (e.g. collection of seed material).
Our knowledge on the genetics of wild olive is very scarce. For instance, we do not know how long-term population fragmentation may have affected the genetic diversity and connectivity among remnant populations. Fragmentation might have resulted into a loss of genetic diversity and led to the formation of genetic differentiation and inbreeding in these metapopulations. It is known that in olive cultivars outcrossing is favored through partial male sterility (Besnard et al. 2000) and that a sporophytic self-incompatibility system is functional (Breton et al. 2014). However, whether the latter is also functional in subsp. cuspidata is not known. The pollen is effectively distributed by wind (Pinillos and Cuevas 2009) and animal-dispersed seeds were on average distributed 190 m in an Ethiopian study (Abiyu 2012). Based on these findings we expected that even in a fragmented landscape wild olive maintains a relatively high level of genetic diversity. To fill these knowledge gaps the main objectives of this study thus were: (i) to quantify genetic diversity in large and small population fragments and among three size classes as a surrogate of age, (ii) to provide estimates of differentiation on the landscape level, (iii) to investigate connectivity and patterns of migration among population fragments, and (iv) to obtain estimates of gene dispersal distances from indirect analysis of gene flow based on SGS. The results we report here are the first on the landscape genetic level for this important Afromontane species.
Materials and methods
Study site
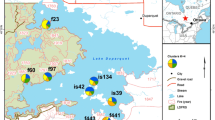
The study was conducted in a natural monastery forest in north-western Ethiopia located 82 km northwest of Bahirdar. Samples were collected in four study plots of different sizes (Table 1). All studied populations are located on hill tops, with only few single trees scattered in the lower lying regions between these population fragments. Most of the habitat between the studied populations is covered by crop and range lands. Plantation forests composed of Eucalyptus and Juniperus trees are found west and north of the largest study site (Tara Gedam, TG). This study site has been under protection of the Tara Gedam monastery since the fifteenth century. This close-to-nature forest has a size of about 130 ha and is highly dominated by O. europaea subsp. cuspidata. The second site is located northwest of TG and composes a relatively large forest (Washa Endrias, WE, ca. 23 ha) where wild olive also dominates and has been similarly protected by the Washa Endrias monastery since the fifteenth century. The remaining two sites (Kidane Mihiret, KM; Sillasie, SL) are located in church forests south of TG and are of very small size (2 and 4 ha, respectively; Table 1; Fig. 1). Hereafter, we treat these study sites as subpopulations and the whole sample as a metapopulation.

Locations of sampled Olea europaea subsp. cuspidata individuals (TG, yellow; WE, blue; KM, Grey and SL, purple; colour of dots denotes the putative source of individuals according to GENECLASS results); arrows denote migration events, numbers next to arrows indicate the number of migrants detected. (Color figure online)
Sample collection, DNA extraction and genetic markers
Leaf samples were taken from a total of 542 trees. In an intensively studied plot (ISP) of 2.53 ha at the core of TG, all existing 307 individuals were sampled. This collection was supplemented by 138 mature individuals randomly selected at TG around this ISP. In KM all existing trees (N = 22) were sampled, while both in WE (N = 48) and SL (N = 27) the samples were randomly selected. Leaves were dried in silica gel and stored at 4 °C until DNA extraction. Diameter at breast height (DBH) as a surrogate of age and spatial coordinates of each tree were recorded. Trees of the ISP were subdivided in three DBH classes (0–20 cm), (21–40 cm), (>41 cm), and thereafter classified as “young”, “medium-aged”, and “old”.
Approximately 1 cm2 of dried leaf sample was crushed in liquid nitrogen and total genomic DNA was extracted using the DNeasy 96 Plant Kit (QIAGEN) according to the manufacturers’ protocol. Ten nuclear microsatellite markers were used for genotyping (Table 2). These markers have been employed successfully in previous studies for genotyping in other subspecies of O. europaea and were selected based on their relatively high level of polymorphism, reliability, and lack of null alleles and genotypic disequilibrium (Besnard et al. 2007).
Multiplex PCR amplifications were optimized to be performed in a 10 μl reaction volume containing 2–10 ng of genomic DNA, 5 μl HotStarTaq Master Mix (Qiagen), double distilled water, and 0.3 µM of forward and reverse primers each. The ten primers used were organized in two multiplexes (primers 1 to 5, and 6–10 from Table 2, respectively). We used the following cycling protocol on a TC-412 Programmable Thermal Controller (Techne): 35 cycles with 94 °C for 30 s, 56 °C for 90 s, and 72 °C for 60 s. Before the first cycle, a prolonged denaturation step (95 °C for 15 min) was included and the last cycle was followed by a 30 min extension at 72 °C. Genotyping was outsourced to a commercial provider (ecogenics, Balgach, Switzerland). For determination of allele sizes on an ABI3730 DNA Analyzer (Applied Biosystems) forward primers were labelled either with Atto565, Atto550, Atto532 (Sigma Aldrich), or FAM (Applied Biosystems) and an internal size standard (LIZ500; Applied Biosystems) was added.
Data analysis
Genetic diversity was analyzed across populations and age classes. Diversity parameters calculated included observed (H o ) and expected heterozygosity (H e ) corrected for different sample size according to Nei (1987), observed (N A ) and effective number of alleles (N e ) according to Nielsen et al. (2003), and fixation index (F is ). The probability for deviation from Hardy–Weinberg expectations (HWE) was assessed after 10,000 permutations, and among population differentiation was assessed by F ST (Weir and Cockerham 1984) and G′ ST (Hedrick 2005). The latter was used as a standarized measure of genetic differentiation, since the high level of polymorphism in microsatellite data can affect the explanatory power of F ST . Allelic richness (A R ) was computed using a rarefaction method for minimizing sampling variances (Petit et al. 1998). All these computations were done using FSTAT v 2.9.3.2 (Goudet 2001), except N e and G′ ST were computed using Genalex v6.5 (Peakall and Smouse 2012). The frequency of null alleles was estimated using the program INEst (Chybicki and Burczyk 2009). This program was also used to calculate the corrected estimates of inbreeding coefficients, as the presence of null alleles can affect F is estimates. INEst was also used to detect potential recent reductions of the population sizes (bottlenecks); here the two-phased model (TPM) of mutation was applied and significance was tested by a Wilcoxon rank test with 10,000 iterations.
A Bayesian model-based clustering analysis at the landscape level was performed using Geneland (Guillot et al. 2005). Individual genotypes and coordinates were used and spatial, non-correlated allele frequencies and the presence of null alleles were assumed during analysis. A total of five independent runs with 100,000 iterations and a burn-in of 200 were set during calculations. In order to explore the genetic connectivity via seed movement, first generation migrants among the four geographically separated subpopulations were analyzed. Migrants towards and out of each subpopulation were identified using GENECLASS2 as individuals which showed less than 5% probability of being a resident (Piry et al. 2004). Assignment of individuals was based on the Paetkau et al. (1995) frequency based criterion and the simulation algorithm described by Paetkau et al. (2004). The proportion of immigrants into a subpopulation was obtained by dividing the total number of immigrants by the total number of sampled individuals in the respective subpopulation.
To get detailed insights into the immigration process in the ISP, the spatial aggregation pattern of immigrants was analysed using the modified Ripley’s–K (L) function (Besag 1977) implemented in SAM 4.0 (Rangel et al. 2010). This test assesses the null hypothesis that immigrants are distributed in complete spatial randomness (CSR). The observed K (L) statistic and the expected value under CSR together with the 95% confidence interval was plotted as a function of the distance radius. A positive K (L) value above the confidence interval indicates a significant clumping of individuals over space in comparison to CSR. To test whether the immigration occurred continuously or discontinuously over generations, the distribution of size (DBH) as a surrogate of age of the immigrants in the ISP was compared with the DBH of residents by using a non-parametric Kolmogorov–Smirnov test (Sokal and Rohlf 1995). This test indicates if the two data sets belong to the same frequency distributions or not. To depict the relative frequency of immigrants compared to residents, the DBH frequency distribution of immigrants and residents in ten size classes were depicted in a histogram.
Spatial genetic structure (SGS) and gene dispersal parameters were estimated using SPAGeDi v1.5 (Hardy and Vekemans 2002). Kinship (ancestral) coefficients according to Loiselle et al. (1995) between individual trees within ten evenly distributed distance class intervals were used to infer SGS. Its significance was determined using the 5% probability value of the upper and lower level confidence intervals obtained after 10,000 permutations of individuals. In this way SGS was assessed for both the ISP and the whole metapopulation, respectively. The regression slope of kinship coefficients to the logarithmic distance b k was used to quantify SGS (Vekemans and Hardy 2004). The significance value of the b k was obtained after 10,000 permutations of individuals. Significant b k indicates the presence of an significant SGS. The Sp statistics which was obtained from −b k /(1−r (1)), where the r (1) refers to the average kinship coefficient of the first distance class was additionally used to quantify SGS. The Sp value allows the quantification of SGS when several populations are compared (Vekemans and Hardy 2004). Calculations of mean gene dispersal distances (α) and neighbourhood size (Nb) were also done in SPAGeDi for the two sampling scales (ISP and whole metapopulation), by an iterative procedure to determine these parameters by regressing pairwise kinship coefficients to logarithmic distance.
Results
In total, 248 alleles (24.8 alleles per locus) were identified (Supplementary Table 1). Observed (H o ) and expected heterozygosities (H e ) were nearly identical for all populations averaging to 0.76 and 0.68, respectively (Table 3). However, null alleles were detected at several loci at a range between 0.00 to 0.35 (Supplementary Table 1), creating heterozygote deficit and leading to departure from HWE in several loci and populations (Supplementary Table 2), and affecting the estimation of inbreeding coefficients (F is ). Therefore F is values per population corrected for the presence of null alleles derived from INest were obtained; these revealed positive and higher F is values for the two smaller subpopulations (SL and KM) compared to subpopulations TG and WE (Table 3). Evenness (N e /A R ) amounted to approximately 0.7 for all populations. In SL and KM recent bottlenecks were likely (p < 0.05). Dividing the core population of TG into three cohorts of different DBH did not result in significantly different estimates, though a weakly significant bottleneck in the young age class was detected (Table 3).
Average subpopulation differentiation was very low (global F ST = 0.016, G′ ST = 0.094; Supplementary Table 2). Pairwise genetic distances were also low (F ST : 0.013–0.042; G′ ST : 0.047–0.158), with WE and SL showing the largest divergence (Table 4). Differentiation measures were also calculated by omitting the two loci with the largest frequency of null alleles (ssrOeUA-DCA11 and UDO099-043), but this did only marginally affect the results (data not shown), therefore all ten loci were retained for the consecutive analyses. The four geographical subpopulations were grouped into two intermingled genetic subpopulations in the Geneland analysis: WE on the one hand and TG, KM and SL on the other hand (Fig. 2). The location of the population fragments on hill tops had no impact on population differentiation; rather spatial distance of WE to the other studied populations appeared as the decisive factor.

Map for the probability of individual membership to two of the putative populations of the metapopulation detected by Geneland. Black dots in the light show Olea europaea subsp. cuspidata individuals from Tara Gedam, Kidane Mihiret and Silassie forests (first cluster) and dots in dark shade show individuals from Washa Endrias (second cluster)
A total of 62 migrants among populations were detected in the GENECLASS analysis. Expectedly, the absolute number of detected immigrants into a geographical subpopulation was dependent on its size, but the proportion of immigrants to the total number of sampled genotypes varied: 7.4% in SL, 10.4% in WE, 11.7% in TG, and 22.7% in KM. Out of the 50 immigrants into TG 64% originated from the two adjacent geographic subpopulations. Migration was mostly bidirectional, except in two cases of unidirectional gene flow (SL to TG; WE to SL; see Fig. 1). Ripley’s K analysis showed that immigrants into the ISP were spatially not randomly distributed but rather clumped (Fig. 3). The Kolmogorov–Smirnov test showed that DBH distribution of immigrants was statistically different (P = 0.04, two-tailed t-test) from the DBH distribution of the non-immigrants. To depict the relative frequency of immigrants compared to residents in the ISP, the relative DBH frequency distribution of immigrants and residents in 10 DBH classes are shown in a histogram (Supplementary Fig. 1). The frequency distribution of DBH classes between immigrants and non-immigrants showed a sharp increase in the fourth class interval (Supplementary Fig. 2).

Results of the modified Ripley’s-K (L) analysis about the spatial aggregation of immigrants of Olea europaea subsp. cuspidata at the ISP: observed values (solid line), theoretical value under CSR (complete spatial randomness; dotted line) and confidence interval (grey area). The observed modified K (L) is above the confidence envelopes at nearly all distances ranges, which indicates that immigrant individuals are spatially significantly clustered at all scales
Kinship coefficients of the first distance class, significant regression slopes (P < 0.001), and the Sp statistics are given in Table 5. For the whole metapopulation up to a distance of about 90 m a significant clumping of similar genotypes was found (Fig. 4). When the ISP was considered alone a significantly positive value was obtained up to a distance of approx. 55 m (Fig. 5). Adults showed slightly stronger spatial structure (Sp = 0.0081) than younger trees (Sp = 0.0064). Gene dispersal parameters were estimated for the metapopulation by assuming an effective population density of 23 individuals per hectare and resulted in a neighborhood size of 1395 trees with a mean gene dispersal (σ) estimate of 220 m. However, no convergence for gene dispersal parameters was obtained when only the ISP at TG was considered.

Correlogram of average kinship coefficient according to Loiselle et al. (1995) (solid line) with 95% confidence interval (dotted lines) to spatial distance (m) of Olea europaea subsp. cuspidata for the whole metapopulation

Correlogram of average kinship coefficient according to Loiselle et al. (1995) (solid line) with 95% confidence interval (dotted lines) to spatial distance (m) of Olea europaea subsp. cuspidata in the intensive study plot
Discussion
High genetic diversity and low genetic differentiation among wild olive subpopulations
Fragmentation of forests in Ethiopia has been ongoing for an extensive time period already. Therefore, comparatively low genetic diversity estimates in small subpopulations due to size and potential reproductive isolation may have been expected. In contrast, our results showed that even relative small geographic subpopulations can maintain high levels of genetic diversity. Heterozygosity estimates were similar among all geographic subpopulations and the average was similar to other studies on wild Olea europaea using microsatellites (Baali-Cherif and Besnard 2005; García-Verdugo et al. 2009; Besnard et al. 2013). We expected a decay of allelic richness in the small geographic subpopulations as this estimate is much more sensitive to population size than heterozygosity. However, allelic richness (after rarefaction) was similar among all subpopulations studied. The relatively high diversity estimates in subpopulations in a fragmented landscape may stem from two reasons: firstly extensive pollen flow via wind and distant seed movement may maintain a high level of genetic diversity even in isolated populations of O. europaea (Baali-Cherif and Besnard 2005; Breton et al. 2006; Besnard et al. 2007, 2009, 2013); secondly, the olive tree is one of the most long-lived tree species (up to thousands of years; Kaniewski et al. 2012) and, therefore, the generation-number related decay of heterozygosity due to drift is to be expected to be relatively small (cf. Baali-Cherif and Besnard 2005). Therefore the current diversity of the populations may indeed also reflect the pre-fragmentation diversity level of a different landscape.
The natural vegetation of the region was dominated by Olea and Juniperus until around the fourteenth to the seventeenth century, and deforestation started afterwards. Darbyshire et al. (2003) showed that the reduction of forest cover in Ethiopian highlands over the past three millennia is highly related to expansion for agricultural land. A reduction of church forests in the study area especially during the past 300 years could have worsened due to the shift of the Axumite dynasty to Gondar and the consecutive expansion of agricultural land and increased demand for fuel wood (Henze 2000). However, the current landscape shape is probably mainly the result of accelerated deforestation in the past 100 years due to intensification of agriculture and increased wood demand (Wassie et al. 2010). The two bigger subpopulations (TG and WE) which were established before the begin of fragmentation, have large population sizes of more than 500 individuals. Therefore, the effect of genetic drift on them is expected to be very small. Theory predicts that at least five generations are required to detect the effect of genetic drift (Young et al. 1996). Taking into account the long life span of the olive tree and the fact that most trees of the studied populations have an adult size, we can assume less than five generations within the past 100 years. Plotting of immigrants in the ISP at TG indicated no direct reduction of seed movement. However, we found a manifestation of the fragmentation in the corrected inbreeding coefficient, i.e. the presence of higher inbreeding in the two smaller subpopulations (SL and KM). In addition, evidence for a recent bottleneck in these populations was provided, although this was not highly significant.
In our investigation very high frequency of null alleles were detected at two of the loci (ssrOeUA-DCA11 and UDO099-043). The corrected per population inbreeding values are much lower than the original estimates in the two bigger populations (TG and WE) and this well explains that most of the increased homozygosity in comparison to the HWE is due to the presence of null alleles. When we divided TG into three cohorts of different ages we expected a higher inbreeding value in the cohort of young trees compared to older trees due to a selection against inbreeds over time (Yazdani et al. 1985; Latouche-Hallê et al. 2003), but we found exclusively significant positive values between 0.01 and 0.02 in all cohorts. Our data contrast findings - F is values were not significantly different from zero - in studies on other O. europaea subspecies which were partly conducted with the same microsatellite loci as used in our study (subsp. laperrinei, Besnard et al. 2007; subsp. guanchica; García-Verdugo et al. 2010).
Subpopulations showed low but significant differentiation (F ST = 0.016; G′ ST = 0.094). This low level of differentiation despite strong fragmentation is probably due to effective pollen dispersal and long distance seed movement by birds and other animals, e.g. civets frequently occur in the study area (Abiyu 2012; Abiyu et al. 2015). In this context it is noteworthy that the pollen-to-seed migration ratio has been found to be close to one in O. europaea subsp. guanchica. This is two orders of magnitude lower than in other wind-pollinated, zoochorous tree species (García-Verdugo et al. 2009). Moreover, one may also argue that our studied subpopulations are the remnants of one formerly larger population and therefore F ST must be small. Although the local scale of our study implies this, results provided by Geneland do not support this argument. Hence, we conclude that effective gene flow very likely hampered a more pronounced subpopulation differentiation as found in other Olea subspecies. Besnard et al. (2009) showed that even four, highly fragmented O. europaea subsp. laperrinei populations in Algeria that were separated by more than 100 km showed very low differentiation (F ST = 0.017) and that effective pollen dispersal could range from less than 100 m to a 2000 m radius depending on the availability of flowering trees, topography and the level of fragmentation. Their global differentiation estimate was almost identical to the one reported in our study. This shows how effective gene flow connects fragmented subpopulations in Olea. For four wild Moroccan and Algerian populations of O. europaea subsp. europaea a slightly higher genetic differentiation was reported [G ST = 0.032, calculated based on data given in Baali-Cherif and Besnard (2005)], while in the Canary Islands subspecies guanchica a much higher genetic differentiation was reported (F ST = 0.15; García-Verdugo et al. 2009).
High immigration from the two small geographical subpopulations (SL and KM) to the biggest one (TG) could be due to frequent dispersal of seeds by civets over limited distance. Abiyu et al. (2015) found high abundance of civetries (defecation sites) inside TG. Presumably civets prefer a dense habitat and/or a home range less affected by humans. Birds are also important dispersers of Olea seeds. Fourteen fruit eating bird species of different size and behavior were identified in TG and in its surroundings (Abiyu 2012). In that study, seeds were predominantly trapped at water holes of a dry river bed located between TG and WE and therefore these water holes may have hampered an even more pronounced seed flow. This highlights how disperser behavior affects local population structure.
The relative proportion of immigrants was highest in KM indicating a sink population. From a source-sink-dynamic point of view a sink population inhabits a relatively poor habitat and might not be viable without the influx of immigrants (Dias 1996). Due to the geographical location of KM this subpopulation might receive more migrants due to frequent bird and civet movements. Moreover, our results indicate that also relative small populations (KM, SL) are effectively contributing to seed flow, highlighting again the fact that even small populations are very important for the conservation of forest genetic resources.
The results of the modified Ripley’s K-function analysis indicated that immigrants were found aggregated across space within the ISP. This might be due to the availability of small forest openings, scattered breeding sites of birds and/or centers of home-ranges of civets. As the DBH distributions of migrants and non-migrants were statistically different, and plotting of the DBH classes for both immigrants and non-immigrants shows no general trend towards decrease or increase of immigration over time, we conclude that immigration occurred erratically rather than synchronously with seeding years. However, the histogram of the diameter distributions (Supplementary Fig. 1) shows a sharp increase in the number of immigrants in the DBH size class 40 which gives an indication of an immigration peak during a certain period.
SGS and gene dispersal
Positive significant SGS in low distance classes and significant negative estimates in high distance classes were found, conforming to an isolation-by-distance (IBD) pattern. SGS slightly varied when different sampling schemes were considered, i.e. at a relatively large spatial scale (landscape level) of up to 3.5 km and at a very fine scale level of less than 300 m. Loiselle’s kinship coefficient in the first distance class amounted to only about 0.01. When we compare this estimate to the coefficients of the same distance class of several neotropical (Hardy et al. 2006) or dipterocarp tree species (Harata et al. 2012), our estimate was very small. SGS is significantly affected by the density of individuals (Doligez et al. 1998), microhabitat heterogeneity (Cruse-Sanders and Hamrick 2004) and local seed shadow overlap (Fuchs and Hamrick 2010); therefore the high number of individuals and the resulting overlap in seed shadows is probably responsible for the low observed kinship values even at the lowest distance class. Similar findings have been reported in other wind polliniated species at high density, e.g. Unger et al. (2011). The estimated mean gene dispersal distance in our study was 220 m. Abiyu (2012) obtained an estimate of seed movement by inverse modeling at TG of 192 m. These two estimates based on very different methodical approaches (seed ecology and populations genetics) are thus nicely coinciding.
At the subpopulations level, we found slightly higher estimates of Loiselles’s kinship coefficients in older trees (Table 5). This slightly stronger spatial structure could be due to differences in seed shadow overlap during colonization of the adult population and mortality of seedlings (Jones and Hubbell 2006). Age-dependent spatial population structure was also found in O. europea subsp. guanchica (old trees F ST = 0.099 vs. young trees F ST = 0.150; García-Verdugo et al. 2009). To a certain proportion we had expected clonal individuals in our subpopulations (cf. Baali-Cherif and Besnard 2005); however, clones were not detected and therefore this seems not to be a common way for reproduction in wild olive.
Conclusions and recommendations to preserve genetic resources
The wild olive has been heavily logged especially for its high quality of charcoal. Additionally illegal grazing by cattle and goats threatens regeneration of this species even in relatively well-protected forests such as TG and WE (see also Zegeye et al. 2011). Moreover, juvenile trees are cut for farm tool preparation and seed picking by local people is further reducing regeneration. These activities diminish the species’ abundance and eventually jeopardize the future survival of wild olive. Therefore, immediate action of protection is required in order to maintain viable populations of this tree species in the future.
Our results of frequent seed movement and low genetic differentiation indicate the relevance of migration in connecting spatially isolated (sub)populations. Predominant sexual reproduction (no clonally reproduced trees were found—unlike in the Saharan laperrinei populations; Besnard et al. 2007), suggests a high potential for adaptation to environmental change. We have shown that even very small remnant populations maintain high genetic diversity. This is an important finding as small spatially up to one km isolated subpopulations can serve as valuable genetic resources. Therefore, conservation and sustainable use of genetic resources should not overlook the value of small populations. At the same time, comparatively high estimates of inbreeding in these small populations in connection with evidence of population bottlenecks, highlights the need for their conservation and defragmentation.
Successful reappearance of wild olive tree in highly degraded areas makes it a key species in rehabilitation of the Afromontane regions in Ethiopia (Bekele 2005). Bekele (2005) and Raf et al. (2006) indicated that recruitment and survival of this tree species highly depends on the availability of moisture and shade. Hence, a screening of genotypes or provenances for drought resistance would increase the success of enrichment plantations. Despite its wide distribution and importance, genetic knowledge of this subspecies is still limited. The current study was undertaken at a landscape level of only a few kilometers radius. Further studies on genetic diversity covering a wider range and further mating system studies in this important tree species are crucial to develop range wide conservation efforts.
References
Abiyu HA (2012) The role of seed dispersal, exclosures, nurse shrubs and trees around churches and farms for restoration of ecosystem diversity and productivity in the Ethiopian highlands. Dissertation, University of Natural Resources and Life sciences, Vienna
Abiyu A, Teketay D, Glatzel G, Gratzer G (2015) Tree seed dispersal by African civets in the Afromontane highlands: too long a latrine to be effective for tree population dynamics. Afr J Ecol 53:588–591
Aerts R, Van Overtveld K, November E, Wassie A, Abiyu A, Demissew S, Daye DD, Giday K, Haile M, TewoldeBerhan S, Teketay D, Teklehaimanot Z, Binggeli P, Deckers J, Friis I, Gratzer G, Hermy M, Heyn M, Honnay O, Paris M, Sterck FJ, Muys B, Bongers F, Healey JR (2016) Conservation of the Ethiopian church forests: threats, opportunities and implications for their management. Sci Total Environ. doi:10.1016/j.scitotenv.2016.02.034
Anderson CD, Epperson BK, Fortin MJ, Holderegger R, James PMA, Rosenberg MS, Scribner KT, Spear S (2010) Considering spatial and temporal scale in landscape-genetic studies of gene flow. Mol Ecol 19:3565–3575
Baali-Cherif D, Besnard G (2005) High genetic diversity and clonal growth in relict populations of Olea europaea subsp. laperrinei (Oleaceae) from Hoggar, Algeria. Ann Bot 96:823–830
Balkenhol N, Cushman S, Storfer A, Waits L (2016) Landscape genetics: concepts, methods, applications, 1st edn. Wiley Blackwell, London
Bekele T (2005) Recruitment, survival and growth of Olea europeaea subsp. cuspidata seedlings and juveniles in dry Afromontane forests of northern Ethiopia. Trop Ecol 46:113–126
Besag J (1977) Contribution to the discussion of Dr. Replay’s paper. J Roy Stat Soc B 39:193–195
Besnard G, Khadari B, Villemur P, Bervillé A (2000) Cytoplasmic male sterility in the olive (Olea europaea L.). Theor Appl Genet 100:1018–1024
Besnard G, Christin PA, Baali-Cherif D, Bouguedoura N, Anthelme F (2007) Spatial genetic structure in the Laperrinei’s olive (Olea europaea subsp. laperrinei), a long living tree from the central Saharan mountains. Heredity 99:649–657
Besnard G, García-Verdugo C, Rubio De Casas R, Treier UA, Galland N, Vargas P (2008) Polyploidy in the olive complex (Olea europaea): evidence from flow cytometry and nuclear microsatellite analyses. Ann Bot 101:25–30
Besnard G, Baali-Cherif D, Bettinelli-Riccardi S, Bouguedoura N (2009) Pollen-mediated gene flow in a highly fragmented landscape: consequences for defining a conservation strategy of the relict Laperrine’s olive. CR Biol 332:662–672
Besnard G, El Bakkali A, Haouane H, Baali-Cherif D, Moukhli A, Khadari B (2013) Population genetics of Mediterranean and Saharan olives: geographic patterns of differentiation and evidence for early generation of admixture. Ann Bot 112:1293–1302
Breton C, Bervillé A (2013) From the olive flower to the drupe: flower types, pollination, self and inter-compatibility and fruit set. In: Barbara S (ed) The Mediterranean genetic code—grapevine and olive, In Tech, pp 291–213
Breton C, Tersac M, Bervillé A (2006) Genetic diversity and gene flow between the wild olive (oleaster, Olea europaea L.) and the olive: several Plio-Pleistocene refuge zones in the Mediterranean basin suggested by simple sequence repeats analysis. J Biogeogr 33:1916–1928
Breton, CM, Farinelli D, Shafiq S, Heslop-Harrison JS, Sedgley M, Bervillé AJ (2014) The self-incompatibility mating system of the olive (Olea europaea L.) functions with dominance between S- alleles. Tree Genet Genom 10:1055–1067
Carriero F, Fontanazza G, Cellini F, Giorio G (2002) Identification of simple sequence repeats (microsatellites) in olive (Olea europaea L.). Theor Appl Genet 104:301–307
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Cipriani G, Marrazzo MT, Marconi R, Cimato A, Testolin R (2002) Microsatellite markers isolated in olive (Olea europaea L.) are suitable for individual fingerprinting and reveal polymorphism within ancient cultivars. Theor Appl Genet 104:223–228
Collevatti RG, Estolano R, Rebeiro ML, Rabelo SG, Lima EJ, Munhoz CBR (2014) High genetic diversity and contrasting fine-scale spatial genetic structure in four seasonally dry tropical forest tree species. Plant Syst Evol 300:1671–1681
Cruse-Sanders JM, Hamrick JL (2004) Spatial and genetic structure within populations of wild American ginseng (Panax quinquefolius L., Araliaceae). J Hered 95:309–321
Cuneo P, Leishman MR (2006) African olive (Olea europeaea subsp. cuspidata) as an environmental weed in Eastern Australia: a review. Cunninghamia 9:545–577
Darbyshire I, Lamb H, Umer M (2003) Forest clearance and regrowth in northern Ethiopia during the last 3000 years. Holocene 13:537–546
De La Rosa R, James CM, Tobutt KR (2002) Isolation and characterization of polymorphic microsatellites in olive (Olea europaea L.) and their transferability to other genera in the Oleaceae. Mol Ecol Notes 2:265–267
Desalegn G, Tadesse W (2010) Major characteristics and potential uses of Eucalyptus timber species grown in Ethiopia. In: Gil L, Tadesse W, Tolosana E, López R (eds) Eucalyptus species management, history, status and trends in Ethiopia. Proceedings of the congress, Ethiopian institute of agricultural research, Addis Ababa, pp 29–52
Dias PC (1996) Sources and sinks in population biology. Trends Ecol Evol 11:326–330
Doligez A, Baril C, Joly HI (1998) Fine-scale spatial genetic structure with nonuniform distribution of individuals. Genetics 148:905–919
Escobar-Páramo P (2000) Microsatellite primers for the wild brown capuchin monkey Cebus apella. Mol Ecol 9:107–118
Fuchs EJ, Hamrick JL (2010) Spatial genetic structure within size classes of the endangered tropical tree Guaiacum sanctum (Zygophyllaceae). Am J Bot 97:1200–1207
García-Verdugo C, Fay MF, Granado-Yela C, Rubio de Casas RR, Balaguer L, Besnard G, Vargas P (2009) Genetic diversity and differentiation processes in the ploidy series of Olea europaea L.: a multiscale approach from subspecies to insular populations. Mol Ecol 18:454–467
García-Verdugo C, Forrest AD, Balaguer L, Fay MF, Vargas P (2010) The relevance of gene flow in metapopulation dynamics of an oceanic island endemic, Olea europaea subsp. guanchica. Evolution 64:3525–3536
Goudet J (2001) FSTAT, version 2.9.3. University of Lausanne. http://www.unil.ch/izea/softwares/fstat.html. Accessed 26 June 2016
Green PS (2002) A revision of Olea L. (Oleaceae). Kew Bull 57:91–140
Guillot G, Mortier F, Estoup A (2005) Geneland: a computer package for landscape genetics. Mol Ecol Notes 5:712–715
Hanski I (1998) Metapopulation dynamics. Nature 396:41–49
Harata T, Nanami S, Yamakura T, Matsuyama S, Chong L, Diway BM, Tan S, Itoh A (2012) Fine-scale spatial genetic structure of ten dipterocarp tree species in a Bornean rain forest. Biotropica 44:586–594
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Maggia L, Bandou E, Breyne P, Caron H, Chevallier M, Doligez A, Dutech C, Kremer A, Latouch-Hallê C, Troispoux V, Veron V, Degen D (2006) Fine-scale genetic structure and gene dispersal inferences in 10 Neotropical tree species. Mol Ecol 15:559–571
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Henze PB (2000) Layers of time: a history of Ethiopia. Hurst and Company, London
Holderegger R, Buehler D, Gugerli F, Manel S (2010) Landscape genetics of plants. Trends Plant Sci 15:675–683
Jones FA, Hubbell SP (2006) Demographic spatial genetic structure of the Neotropical tree, Jacaranda capaia. Mol Ecol 15:3205–3217
Jordano P, Garcia C, Godoy JA, Garcia-Castano JL (2007) Differential contribution of frugivores to complex seed dispersal patterns. Proc Natl Acad Sci 104:3278–3282
Kaniewski D, Boiy T, Terral JF, Khadari B, Besnard G (2012) Primary domestication and early uses of the emblematic olive tree: palaeobotanical, historical and molecular evidence from the Middle East. Biol Rev 87:885–899
Keller D, Holderegger R, Van Strien MJ, Bolliger J (2015) How to make landscape genetics beneficial for conservation management? Conserv Genet 16:503–512
Latouche-Hallê C, Ramboer A, Bandou E, Caron H, Kremer A (2003) Nuclear and chloroplast genetic structure indicate fine-scale spatial dynamics in a Neotropical tree population. Heredity 91:181–190
Loiselle BE, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Mills LS, Allendorf FW (1996) The one-migrant-per-generation rule in conservation and management. Conserv Biol 10:1509–1518
Nathan R, Schurr FM, Spiegel O, Steinitz O, Trakhtenbrot A, Tsoar A (2008) Mechanisms of long-distance seed dispersal. Trends Ecol Evol 23:638–647
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nielsen R, Tarpy DR, Reeve HK (2003) Estimating effective paternity number in social insects and the effective number of alleles in a population. Mol Ecol 12:3157–3164
Oddou-Muratorio S, Demesure-Musch B, Pélissier R, Gouyon PH (2004) Impact of gene flow and logging history on the local genetic structure of a scattered tree species, Sorbus torminalis L. Crantz. Mol Ecol 13:3689–3702
Paetkau D, Calvert W, Stirling I, Strobeck C (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol Ecol 4:347–354
Paetkau D, Slade R, Burdens M, Estoup A (2004) Genetic assignment methods for the direct real-time estimation of migration rate: a simulation based exploration of accuracy and power. Mol Ecol 13:55–65
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-un updatae. Bioinformatics 28:2537–2539
Petit RJ, El Mousadik A, Pons O (1998) Identifying populations for conservation on the basis of genetic markers. Conserv Biol 12:844–855
Pinillos V, Cuevas J (2009) Open-pollination provides sufficient level of cross-pollen in Spanish monovarietal olive orchards. Hortscience 44:499–502
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Raf A, Eva N, Ives VB, Mintesinot B, Martin H, Bart M (2006) Effects of pioneer shrubs on the recruitment of the fleshy-fruited tree Olea europaea ssp. cuspidata in Afromontane savanna. Appl Veg Sci 9:117–126
Rangel TF, Diniz-Filho JAF, Bini LM (2010) SAM: a comprehensive application for spatial analysis in macroecology. Ecography 33:46–50
Sefc KM, Lopes MS, Mendonça D, Dos santos MR, Da Cámara Machado ML, Da Cámara Machado A (2000) Identification microsatellite loci in olive (Olea europaea) and their characterization in Italian and Iberian olive trees. Mol Ecol 9:1171–1193
Sokal RR, Rohlf FJ (1995) Biometry: the principles and practices of statistics in biological research, 3rd edn. WH Freeman and Company, New York
Unger GM, Konrad H, Geburek T (2011) Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst Evol 292:133–141
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Wang R, Compton SG, Chen XY (2011) Fragmentation can increase spatial genetic structure without decreasing pollen-mediated gene flow in a wind-pollinated tree. Mol Ecol 20:4421–4432
Wassie A, Sterck FJ, Bongers F (2010) Species and structural diversity of church forests in a fragmented Ethiopian highland landscape. J Veg Sci 21:938–948
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Yazdani R, Muona O, Rudin D, Szmidt AE (1985) Genetic structure of a Pinus sylvestris L. seed-tree stand and naturally regenerated understory. Forest Sci 7:430–436
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Zegeye H, Teketay D, Kelbessa E (2011) Diversity and regeneration status of woody species in Tara Gedam and Abebaye forests, northwestern Ethiopia. J Forest Res 22:315–328
Acknowledgements
We would like to acknowledge the project “Carbon storage and soil biodiversity in forest landscapes in Ethiopia: Knowledge base and participatory management (Carbo part)” for supporting this work. We also thank C. Dobeš and J.P. George for commenting on the manuscript, T. Thalmayr for help with figures, and D. Fentie for assisting in sample collection.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kassa, A., Konrad, H. & Geburek, T. Landscape genetic structure of Olea europaea subsp. cuspidata in Ethiopian highland forest fragments. Conserv Genet 18, 1463–1474 (2017). https://doi.org/10.1007/s10592-017-0993-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0993-z