
Abstract
The genetic effects of harvest may be especially important in species that form social groups, such as gray wolves (Canis lupus). Though much research exists on the ecology and population dynamics of gray wolves, little research has focused on how anthropogenic harvest relates to the genetics of wolf populations. To analyze the short-term genetic consequences of the first two years of public wolf harvest in Minnesota following delisting under the Endangered Species Act, we genotyped harvested individuals at 18 microsatellite loci and quantified changes in population genetic structure and diversity in the first post-harvest year. If the harvest rate was high enough to create detectable genetic changes, population structure and differentiation between clusters could both increase because of decreased natal dispersal and increased disperser mortality, or they could decrease because of increased immigration from outside the population. In the Minnesota population, heterozygosity and allelic richness were not significantly different between years. However, population genetic structure increased and effective migration decreased among the sampled wolves. While the role of anthropogenic harvest in these changes cannot be distinguished from other confounding factors, this analysis suggests that harvest has a non-negligible effect and indicates the need for continued study to determine whether harvest-induced changes in genetic structure affect the evolutionary trajectory of harvested populations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Understanding how human harvest affects wild populations is important for maintaining consistency between hunting and trapping regulations and long-term conservation goals. Science-based harvest management is often effective at regulating population sizes and reducing human-wildlife conflicts, but excessive harvest can lead to reduced genetic diversity or altered population structure (Allendorf et al. 2008). Specifically, excessive harvest can substantially reduce a species’ ability to respond to environmental change or anthropogenic disturbances and alter a species’ evolutionary trajectory (Coltman 2008; Frankham 2005).
Harvest may drive evolution via genetic drift, natural selection, or gene flow. As population size decreases, loss of genetic variation in a population accelerates (Reed and Frankham 2003; Frankham 2005) and the impact of genetic drift and potential for inbreeding depression both increase (Frankel and Soulé 1981). Selective harvest alters natural selection if particular phenotypes and their underlying genotypes are favored. This can lead to demographic and phenotypic shifts in the population, such as earlier age at maturation in Atlantic cod and other fishes (Kuparinen and Merilä 2007), decreased horn sizes in trophy ungulates (Coltman et al. 2003), and decreased size and weight at sexual maturity in salmonid fishes (Hard et al. 2008). Hunting could also drive natural selection by (a) reducing the overall fitness of the population if the fittest individuals are removed, (b) improving population fitness by removing the weakest individuals, or (c) changing the fitness landscape by making harvest-avoidance strategies selectively advantageous. The selective role of harvest and its genetic consequences in wild populations have been explored and reviewed extensively (e.g. Harris et al. 2002; Frankham 2005; Fenberg and Roy 2008; Milner et al. 2007; Allendorf and Hard 2009).
Finally, harvest can significantly affect dispersal and gene flow between populations. Dispersal generally increases fitness when habitat quality is heterogeneous in time and space (Olivieri et al. 1995; Ronce 2007), reduces kin competition (Hamilton and May 1977; Frank 1986; Ronce 2007), and reduces inbreeding (Greenwood et al. 1978; Waser et al. 1986). Dispersal plays an essential role in maintaining genetic diversity in many species and can aid in the genetic recovery of a population from harvest (Allendorf et al. 2008). However, these advantages must outweigh the disadvantages of dispersal, which may include greater energy expenditure, increased predation, reduced survival or reproductive success in a new environment, and increased competition in a new environment (Garant et al. 2007). In cooperative breeders, four factors mediate an individual’s choice to delay dispersal: the benefits gained by remaining in its natal group, its relatedness to other individuals in the group, whether the individual is tolerated by the dominant members of the group (e.g. its parents), and constraints that limit opportunities for establishing a territory and reproducing on its own (Koenig et al. 2016). Due to the interplay between these factors, dispersal behavior is often plastic, with individual organisms modifying their dispersal behavior (timing, distance, and direction) in response to biotic and abiotic changes (Garant et al. 2007; Ronce 2007).
Gray wolves (Canis lupus) live in family-based social groups and depend on dispersal primarily for inbreeding avoidance and reduced kin competition (Mech and Boitani 2003). Packs are territorial and generally contain a dominant unrelated breeding pair and their offspring from multiple years (Mech 1999). Juvenile wolves of both sexes typically disperse from their natal packs following sexual maturity (Gese and Mech 1991) to establish their own packs, with most wolves dispersing between 11 and 24 months of age (Mech and Boitani 2003). However, juveniles will remain in the natal pack longer if resources allow and lower dispersal rates have been documented in areas or time periods with higher ungulate densities (Ballard et al. 1987; Gese and Mech 1991). Here, we consider pups to be wolves <1 year of age, and these individuals are expected to still live in their natal packs. Juveniles are the dispersers, between 1 and 2 years of age, and wolves >2 years of age are considered adults.
At low harvest rates, wolf packs and populations compensate for harvest-related mortality through changes in recruitment rates (Ballard et al. 1987; Sparkman et al. 2011) and dispersal behavior (Adams et al. 2008). High levels of harvest make dispersal riskier, reducing emigration rates, dispersal distances, and survival of dispersers from harvested areas (McCullough 1996; Newby et al. 2013). This decreases gene flow between populations (Harris et al. 2002) and potentially reduces population resilience (Adams et al. 2008). In addition, weaker intraspecific competition in harvested areas may lead to increased immigration from unharvested populations (Adams et al. 2008; Webb et al. 2011). This creates a source-sink dynamic between populations: areas with high harvest rates become sink populations as individuals from lightly harvested populations disperse into those areas (Andreasen et al. 2012; Newby et al. 2013). Each of these factors may impact a population’s genetic structure as a result of anthropogenic harvest.
Unharvested packs generally exhibit high intra-pack relatedness and are more closely related to nearby packs than those farther away (Wayne and Vilà 2003), resulting in a pattern of isolation by distance (IBD; Rutledge et al. 2010). Spatially heterogeneous reductions in wolf density can increase immigration into and/or decrease emigration from an area (Adams et al. 2008) by relaxing resource competition. Unrelated juvenile dispersers may be ‘adopted’ into packs subject to harvest (Lehman et al. 1992; Grewal et al. 2004; Jędrzejewski et al. 2005), reducing average relatedness and increasing genetic variability within packs. Territory turnover resulting from breeder turnover and pack dissolution at high levels of harvest can lead to greater genetic homogenization among wolves across the harvested population (Brainerd et al. 2008; Rich et al. 2012; Caniglia et al. 2014). Such increased movement of individuals between packs in heavily harvested populations and recruitment of outside wolves into the harvested population may increase population-wide genetic homogeneity and reduce genetic structure (Jędrzejewski et al. 2005). If this is the case, a breakdown of IBD, decrease in genetic structure, and increase in genetic diversity should be detectable in a newly harvested wolf population. Alternatively, reduced dispersal rates from harvested areas due to low wolf densities and increased disperser mortality could reduce gene flow between subpopulations, which would result in increased differentiation between subpopulations and reduced genetic diversity. Though much energy has been put into research on the ecology, social structure, and dynamics of wolf populations (reviewed in Fritts et al. 2003; Boitani 2003), little research has focused on the total effect of anthropogenic harvest on wolves and how this relates to the genetics of wolf populations.
In this study, we analyzed genetic diversity and population structure before and after the onset of harvest-based wolf management in Minnesota. Wolves were protected under the Endangered Species Act (ESA) in the contiguous United States from 1974 to 2012, during which time anthropogenic wolf mortality from legal removal following wolf depredation on livestock or pets occurred, as well as an unknown amount of illegal harvest. In Minnesota, depredation control resulted in the mortality of 5–8% of the estimated population per year between 1988 and 2012 (USFWS Twin Cities Ecological Services Field Office 2014). The Great Lakes Distinct Population Segment (including Minnesota, Wisconsin, and Michigan) was delisted under the ESA in 2012, returning wolf management to state agencies. Public harvest in Minnesota in 2012 resulted in the hunting and trapping of 413 wolves, in addition to 296 wolves killed for depredation control. This was the highest recorded anthropogenic mortality rate of wolves in Minnesota in recent history (approximately 24% of the estimated adjusted population size; Fig. S1).
Fuller (1989) predicted that harvest rates up to 28% of the Minnesota wolf population should be sustainable. However, even with a sustainable harvest, there may be genetic consequences for a population, such as changes in gene flow or genetic structure, which may be evident even within one generation. We hypothesized that if the realized anthropogenic mortality rate of 24% was genetically neutral for the Minnesota wolf population, then we would see no population-wide change in genetic indices and population structure between samples collected prior to legal harvest and the population sampled after harvest. If the harvest rate was high enough to create detectable genetic changes, we anticipate that either (a) population structure and differentiation between clusters both increased because of decreased natal dispersal and increased disperser mortality; or (b) population structure decreased and genetic diversity and homogeneity increased resulting from an increase in immigration from outside the population.
We tested our hypotheses using hunter-harvested samples from a “pre-harvest” and “post-harvest” population, focusing primarily on genetic diversity, gene flow, and population structure. We analyzed changes in genetic diversity, effective population size, and spatial population genetic structure of wolves in Minnesota over this two-year sampling period to provide insight into the short-term effects of increased anthropogenic mortality on dispersal and gene flow in harvested wolf populations.
Methods
Samples and study area
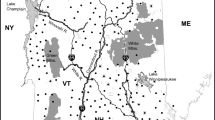
Muscle tissue samples were collected from legally harvested wolves during carcass inspection by the Minnesota Department of Natural Resources (MNDNR) during the 2012–2013 (“pre-harvest”, n = 413) and 2013–2014 (“post-harvest”, n = 238) wolf harvest seasons. Harvest was lower in the second year of sampling due to regulation changes. We considered the first year of harvest a “pre-harvest” sample, because the anthropogenic mortality rate prior to the first legal harvest (including depredation control and illegal harvest) was relatively low and constant over the previous 30-year period. Hunters and trappers reported the county, township, range, and section of harvest. We determined the age of all individuals using cementum annuli counts (Stark and Erb 2013). Harvest sex ratios were approximately 50:50 in both years (Stark and Erb 2013, 2014). Juveniles typically represent the single largest cohort in Minnesota wolves (Mech 2006), and this is reflected in the age distribution of harvested wolves in both years (Stark and Erb 2013, 2014). Minnesota wolf range encompasses most of the forested portion of the state, and harvest samples were distributed throughout this area in both years (Fig. 1).

Sampling locations for pre-harvest wolf samples (dark circles, n = 413) and post-harvest samples (light circles, n = 238) used in this study. The shaded area represents the current estimated occupied wolf range in Minnesota, adapted from Erb and Sampson (2013). (Color figure online)
Laboratory methods
We extracted DNA from muscle tissue samples using the GeneJET Genomic DNA Purification Kit (ThermoScientific, Waltham, MA, USA). Extractions followed kit protocol, with modifications to improve DNA concentration and purity. We quantified DNA and diluted it for downstream analyses. We PCR-amplified one Y-chromosome locus (SRY; Meyers-Wallen et al. 1995) and two X-chromosome microsatellites (FH2548, FH2584; Richman et al. 2001) in a multiplex reaction to confirm extraction success and determine the sex of each sample. We scored results based on presence of the X-chromosome bands (for extraction success) and the presence of an SRY band (for sex determination) using agarose gel electrophoresis. We extracted additional muscle tissue for individuals that did not have positive DNA quantification or sex-determination results, and omitted samples with two failed extractions.
We then amplified the DNA at 20 autosomal microsatellite loci and two x-linked microsatellites used previously in C. lupus studies, based on C. lupus familiaris microsatellites (Table S1). These included 14 tetranucleotide repeat loci (FH2054, FH2422, Breen et al. 2001; FH3313, FH3725, FH3853; Guyon et al. 2003; FH2004, FH2611, FH2785, FH3965; Francisco et al. 1996; PEZ06, PEZ08, PEZ11, PEZ12, PEZ15, J. Halverson in; Neff et al. 1999) and six dinucleotide repeat loci (CPH3, Fredholm and Winterø 1996; C05.377, C10.213; Ostrander et al. 1993; Ren106I06, Ren169O18, Ren239K24; Breen et al. 2001). These 20 autosomal microsatellites represent 17 of the 38 total C. lupus chromosomes, with only three locus pairs on the same chromosomes (C10.213 and FH2422 on CFA10, Ren169O18 and Ren239K24 on CFA29, PEZ15 and C05.377 on CFA5; Neff et al. 1999). The two x-chromosome tetranucleotide repeat loci were FH2548 and FH2584 (Richman et al. 2001). Loci FH3965, PEZ15, FH2054, and C10.213 were later removed from analysis due to low amplification success, resulting in 18-locus genotypes for downstream analyses.
We amplified microsatellites in a combination of single and multiplex reactions (Table S1) using M13-labeled forward primers (Schuelke 2000). Each reaction contained 2 ng of template genomic DNA, 1X GoTaq Flexi buffer, 2 mM MgCl2, 0.2 mM dNTPs, 0.5 ng/µl bovine serum albumin, 0.08 µM unlabeled forward primer, 0.8 µM reverse primer, 0.8 µM M13-labeled fluorescent primer, GoTaq DNA polymerase (Promega, Madison, WI, USA), and sterile water to reach 13 µl total volume. We PCR-amplified products using 2 min denaturation at 95 °C followed by 30 amplification cycles of 30 s at 95 °C, 30 s at the locus- or multiplex-specific annealing temperature (Table S1), and 1 min at 72 °C, and with a final 10 min extension at 72 °C. We combined PCR products into genotyping panels (Table S1), which were analyzed on an ABI 3730xl capillary genetic analyzer at the University of Minnesota Biomedical Genomics Center. We scored alleles using Genemarker (v.2.6.0, SoftGenetics LLC) and amplified uncertain alleles a second time to confirm genotypes.
Error checking
To estimate accuracy in genotyping results, five or more randomly chosen individuals were re-amplified at each locus. We calculated the presence and frequency of microsatellite errors such as allelic dropout, false alleles, and null alleles using Microchecker (Van Oosterhout et al. 2004). We also estimated null allele frequency for each locus and polymorphic information content (PIC) using Cervus (Kalinowski et al. 2007).
Genetic diversity and inbreeding.
We estimated observed (Ho) and expected (He) heterozygosities and the number of alleles (NA) in Arlequin (v3.5, Excoffier and Lischer 2010). We used Fstat (Goudet 1995) to calculate allelic richness (AR) and fixation index (FIS) for each locus individually and each sampling year as a whole. We used the linkage disequilibrium method in NeEstimator (v2, Do et al. 2014) to estimate effective population sizes for each sampling year assuming monogamy and excluding alleles with a frequency <0.01. Though the assumption of monogamy rather than random mating can inflate estimates of Ne in a population, we were particularly interested in the relative Ne in pre-harvest and post-harvest populations, and using Ne as a measure of the strength of drift as an evolutionary force acting on the population, rather than absolute Ne for each year. While wolves are not strictly monogamous, the breeding pair generally remains together unless one of the pair dies (Milleret et al. 2016) and not every wolf breeds in a population, and thus we chose to assume monogamy.
Population structure
We used Bayesian genotype assignment in Structure (v2.3, Pritchard et al. 2000) to infer the most probable number of genetically distinct clusters and to estimate admixture proportions within each of those clusters. In Structure, genotypes are assigned a probability of membership (Q) in each of K genetic clusters, where K represents the a priori specified number of genetic subpopulations. We ran the admixture model of Structure assuming correlated allele frequencies from K = 1 to K = 10, three times each for 500,000 iterations following a 50,000 iteration burn-in for each of the two sampling years. Due to uneven sample sizes between the 2 years, we subsampled the pre-harvest sample set by (1) removing those individuals with the most missing data, followed by (2) selecting the nearest individual without replacement for each post-harvest sample, resulting in a geographically paired subsample set. We used a combination of inspecting the log-likelihood of the probability of the data (lnP(D)) for each value of K and the ∆K method (Evanno et al. 2005), implemented in Structure Harvester (Earl and VonHoldt 2012), to determine the most probable number of clusters K for each sampling year. We then ran Structure again at the most probable K value for 10 runs, from which the individual assignment probabilities were averaged using Clumpp (Jakobsson and Rosenberg 2007) to obtain final values (Q) of cluster assignment and admixture for each individual. We categorized individuals with any Q i ≥ 0.6 as belonging to cluster i, and those with all Q i < 0.6 as admixed. We used FST and admixture (α) values to characterize differentiation between Structure-produced clusters, and determined significance via permutation in Fstat (Goudet 1995).
Structure can overestimate genetic divisions in populations displaying a pattern of genetic IBD if IBD is not accounted for (Frantz et al. 2009; Schwartz and McKelvey 2009). To test for IBD, we used Mantel tests of the correlation between log-transformed Euclidean geographic distance and linear genetic distance (â, Rousset 2008) between each pair of individuals in Arlequin (v3.5, Excoffier and Lischer 2010). Rousset’s â genetic distance is appropriate for individuals at a small scale in a continuously distributed population (Rousset 2000) and thus was chosen for our analyses.
In addition to Mantel tests to quantitatively determine whether genetic distances could be explained by IBD, we used spatial structure analysis in GenAlEx (v6.501, Peakall and Smouse 2012) at 50 km distance categories, with 999 permutations and 9999 bootstraps. Spatial structure analysis computes the correlation coefficient between genetic distance (Rousset’s â, calculated in Genepop) and geographic distance for individuals within certain geographic distance bins, to determine whether the correlation changes as the geographic distance changes (Smouse and Peakall 1999). We expected wolves within the distance of a pack territory (~15–20 km diameter) to be highly related to one another, with decreasing relatedness at larger distances.
Geographic distribution of clusters
The locations reported by hunters to the MNDNR upon wolf carcass inspection were mapped to the centroid of the reported section. These locations represent the locations where wolves were harvested, rather than known pack locations, which introduces uncertainty into the analysis due to the highly mobile nature of wolves [individuals may travel 14 km from one side of their territory to the other (Erb et al. 2014) and may disperse hundreds of kilometers (Fritts 1983)]. The mapped locations are accurate to the harvest location within the distance of a section (1.1 km). Population cores for Structure-defined clusters were visualized by computing “hotspots” for each cluster using Getis-Ord G* hot spot analysis (Getis and Ord 1992) in ArcMap (v.10.2.2, ESRI), based on Q-values from Structure. Minimum convex polygons (MCP) were drawn around all of the sample points with 95% hotspot significance (to delineate the 95% core) and 99% hotspot significance (to delineate the 99% core) for each cluster.
To visualize spatial patterns of gene flow, we used the individual-based estimated effective migration surfaces (EEMS) program (Petkova et al. 2014). Cluster-based methods of determining gene flow have traditionally been used in genetic structure analyses, but these are not well suited for use in populations with continuous patterns of genetic variation, such as those exhibiting IBD (Rousset 2000). EEMS models gene flow across a landscape as a function of individual-based migration rates (Petkova et al. 2014), calculated using a stepping stone model (Kimura and Weiss 1964). Interpolated migration surfaces delineate areas with higher than expected estimated migration rates (i.e. corridors for gene flow) and those with lower than expected estimated migration rates (i.e. barriers to gene flow). We averaged three runs each of 5,000,000 MCMC iterations (sampled every 1000 steps following a 1,000,000 step burn-in) with 50, 150, 200, 300, and 400 demes to produce the final EEMS surfaces. We chose this range of demes because the 400-deme (170 km2 deme size) grid falls within the range of average wolf pack territory size for Minnesota (78–260 km2; Fuller 1989; Fuller et al. 1992; Erb et al. 2014), while the 50-deme grid (1800 km2 deme size) can detect processes occurring on a larger scale.
Historical migration rates
We used Migrate (v3.6, Beerli and Palczewski 2010) to estimate the historical migration rates between Structure-defined clusters in the Minnesota wolf population. In Migrate, the single step stepwise mutation model was chosen with a threshold of 100 bp (maximum 100 bp difference between alleles at a given locus), with FST used as the start parameter for population mutation rate (θ) and migration rate. A Bayesian Markov Chain Monte Carlo (MCMC) search strategy was used with 500,000 steps sampled following 100,000 burn-in steps.
Results
Microsatellite genotyping and genetic diversity
A total of 634 samples (401 from pre-harvest, 233 from post-harvest) were successfully amplified at 16 or more microsatellite loci and included in analyses. Individuals with fewer than 16 successful loci were not included in any analyses. All 18 microsatellite loci were polymorphic in both sample years. Error analysis indicated the presence of null alleles in Ren106I06, PEZ11, FH2004, and PEZ08, with an average null allele frequency of 0.047 (SE = 0.011). Expected heterozygosity (He) for loci ranged from 0.59 to 0.92 and observed heterozygosity (Ho) ranged from 0.29 to 0.88 (Table S1), indicating high overall genetic diversity in the population. The combined set of samples had He and Ho that were not significantly different from one another (Table 1; He = 0.82 ± 0.08, Ho = 0.72 ± 0.15).
In comparing genetic diversity indices between years, we found no significant differences. The estimated effective population size was slightly but not significantly lower in the post-harvest samples than in the pre-harvest samples, and other measures of genetic variation also indicate a slight but generally non-significant decrease in variation from pre-harvest to post-harvest (Table 1).
Population genetic structure
Based on the subsampled data set, the most probable number of clusters K for the pre-harvest sample was 3, while the most probable number of post-harvest clusters was 4. Inspection of posterior probabilities and deltaK plots (Evanno et al. 2005) for both years supported these inferences (Fig. S2, S3). In the pre-harvest year, individuals were roughly equally split among the three clusters (30, 22, and 30%) and 16% did not have any clustering assignment Q i > 0.6 (Fig. 2). The admixture coefficient (α) was 0.10, average FST = 0.029, and the mean sample size-corrected number of migrants between clusters per generation (Nm) calculated using the private alleles method was 8.2 (Table 2). Post-harvest, individuals were less equally split (23, 19, 11, and 20%) and 27% were considered admixed (Fig. 2). These post-harvest clusters had α = 0.096, mean FST = 0.043, and a mean Nm of 7.2 individuals per generation. These three pre-harvest and four post-harvest clusters were used for all downstream cluster-based analyses.

Bayesian clustering assignments at K = 3 from Structure for n = 233 wolves from the pre-harvest sample and at K = 4 for the post-harvest sampling year. In both graphs, each vertical bar corresponds to an individual wolf, and the colors of each bar correspond proportionally to that individual’s probability of cluster membership to the south-central (blue), northeast (green), northwest (yellow), and north-central (red) clusters. Samples are separated by cluster assignment, with unassigned (admixed) individuals as the final group. Individuals were assigned to clusters using a cutoff value of 60% (Q = 0.6). (Color figure online)
In both years, the Getis-Ord G* hot spot analysis of the locations produced population cores generally corresponding to the south-central (SC, cluster 1), northeast (NE, cluster 2), and northwest (NW, cluster 3) areas of wolf range in Minnesota, with an additional north-central (NC, cluster 4) cluster in the post-harvest year (Fig. 3a, c). Pre-harvest, the core areas of the three clusters (using α = 0.01) were generally mutually exclusive, with the NE core smallest and NW core largest (Fig. 3c). The cores at α = 0.05, however, overlap in the central part of wolf range, with the SC core extending across much of the southern region. Post-harvest, the cores were similar to one another in size (excepting the small SC core) and shaped differently than the pre-harvest cores (Fig. 3d). The cores at α = 0.05 overlap less than they do pre-harvest, supporting the FST and Nm results suggesting increased differentiation and decreased migration between post-harvest clusters when compared to the pre-harvest sample (Table 3).

Spatial analysis of Structure-assigned Bayesian clustering results. a Maps of individuals assigned to each of the three clusters, and admixed individuals (Ad) with no cluster assignment, from pre-harvest samples. b Getis-Ord G* Hotspot analysis cores (solid: significant at 99% confidence, hollow: 95% confidence) for the three populations in the pre-harvest year, based on Q-values derived from Structure analyses. c Locations for individuals assigned to each of the clusters, and admixed individuals, for the post-harvest samples. d Hotspot analysis-derived cores for the post-harvest sampling year. (Color figure online)
Geographic distribution of clusters
Mantel tests were used to determine whether there was a significant correlation between genetic distance (â) and the log-transformed geographic distance between individuals. The pre-harvest and post-harvest sample years were both significantly geographically structured, such that genetic distance could be explained at least in part by geographic distance (Table 4; p < 0.001). However, this correlation explained <1% of the variation in both the pre- and post-harvest samples. All three pre-harvest clusters also had significant correlations between genetic distance and geographic distance (all p < 0.01), and the same was true for the four post-harvest clusters (all p < 0.05). All correlation coefficients (r) in both years were less than 0.28.
This difference in genetic structuring with distance was also evident in the spatial structure analyses for pre- and post-harvest. In the pre-harvest samples, genetic distance and geographic distance were correlated for the combined sample set at close distances and the correlation weakened with increasing distance (Fig. S4), indicating IBD within the first two distance classes and a breakdown of IBD for samples in bins greater than 50 km (intercept: 98 km). The correlograms for spatial structure analysis were significantly different from r = 0 in the pre-harvest sample set as a whole (p = 0.001), as well as in the northeast (p = 0.02) and northwest (p = 0.001) populations individually, but only marginally significant in the south-central population (p = 0.05). In the post-harvest sampling year, the sample set as a whole and all four Structure-defined clusters had individual correlograms that were significantly different from the null hypothesis of no correlation (p < 0.001), although the r values were smaller than pre-harvest (maximum r = 0.021 ± 0.003; Fig. S4), indicating weaker correlation between genetic and geographic distances. The post-harvest sample set as a whole had an intercept of 91 km, which was not significantly different from the pre-harvest intercept. However, the average x-intercept of the four post-harvest clusters separately was 42.1 km (s.d. = 3.8), while the average x-intercept of the three pre-harvest clusters separately was 56.3 km (s.d. = 21.8). This suggests that IBD breaks down at a smaller distance following harvest than it did before harvest began.
Population bottlenecks and historical gene flow
We produced individual-based effective migration surfaces for each year using EEMS (Fig. 4). Using the sub-sampled pre-harvest samples, there is an area of high resistance to gene flow in the west-central portion of wolf range in Minnesota (Fig. 4). In addition, a resistive band runs east to west across wolf range, generally following Highway 2 across the state. Gene flow corridors lie in the northeast, south, and northwest parts of wolf range in the state, roughly corresponding to the core areas for each of the three Structure-defined clusters. Following harvest, the effective migration surface has a larger proportion of resistive (orange-colored) areas than in the previous year (Fig. 4). As with the pre-harvest sampling year, the post-harvest resistance surface has pockets of enhanced gene flow in the northeast, south, and northwest parts of wolf range in the state, roughly corresponding to the core areas for the Structure-defined clusters. The larger patch of resistive surface between the northwest and north-central portions of the state following harvest corresponds to where the fourth cluster (NC) is delineated following harvest.

Individual-based EEMS analysis of effective migration rates (m) for the pre-harvest and post-harvest samples. Colored regions indicate areas that deviate from isolation by distance (IBD). Darker orange areas have lower than expected effective gene flow, while darker blue areas have higher than expected gene flow (i.e. are corridors to gene flow). Migration surfaces are averages of three runs each with 50, 150, 200, 300, and 400 demes. (Color figure online)
Historical maximum likelihood migration rates, estimated in Migrate using the full post-harvest sample set, show net effective migration predominantly from the northeast cluster toward the northwest and south-central clusters (Fig. 5). Modal mutation-scaled migration rates in number of migrants per generation were highest leaving the NE cluster (MNE−SC = 7.28, MNE−NW = 16.49) and lowest entering the NE cluster (MSC−NE = 0.0065, MNW−NE = 0.012). Estimated migration rates between the NW and SC clusters were similar to or slightly lower than the emigration rates from the NE cluster (MNW−SC = 4.02, MSC−NW = 7.83). This directionality of historical migration shows wolves dispersing from the interior of the range toward the edge of known wolf range in Minnesota.

Historical net migration rates between pre-harvest Structure-defined clusters in Minnesota, as estimated using Migrate. Values are mutation-scaled net migration rates in numbers of migrants between clusters per generation, with directionality of net migration indicated by the arrows
Discussion
In this study, we used two consecutive years of harvest samples from across wolf range in northern Minnesota to determine whether harvest had detectable short-term impacts on population genetic structure and patterns of gene flow across the state. Both sampling years had high genetic diversity and a slight deficiency in heterozygotes, which is common in wolves due to their non-random breeding patterns and social structure (Wayne and Vilà 2003). The estimated effective population size did not change between years, suggesting that genetic drift was not a significantly stronger force in the first year post-harvest than it was in the time period leading up to harvest. There was evidence for reduced local IBD, reduced gene flow, and increased differentiation between clusters in the post-harvest (2013–2014) sample set when compared to pre-harvest (2012–2013), suggesting that more individual wolves remained in or near their natal packs following harvest. Low disperser survival and reduced local resource competition due to harvest may have both contributed to the observed reduction in gene flow. In cooperative breeders, such delayed dispersal often increases breeding opportunities (Koenig et al. 1992) and survival (via increased foraging efficiency and protection from predation; Alexander 1974), which both increase individual fitness (Jennions and Macdonald 1994).
A small portion of the differentiation in the pre-harvest samples could be explained by IBD (average r = 0.14), whereas the post-harvest clusters had a weaker association between genetic and geographic distances (average r = 0.12; Table 4). Though weakly significant, this change is also evident in the spatial structure differences between pre- and post-harvest (Fig. S4). We believe that this reduced IBD is indicative of increased homogenization at a local scale. Local (within-cluster) genetic structure would decline following a disruption of the normal pack composition and territorial mosaic, phenomena which have been correlated with increased anthropogenic mortality in wolves (Brainerd et al. 2008). Previous studies have found that more unrelated wolves joined harvested packs than unharvested packs in Ontario (Grewal et al. 2004) and Poland (Jędrzejewski et al. 2005), reducing intra-pack relatedness. Increased anthropogenic mortality is also associated with increased rates of pack dissolution (Brainerd et al. 2008). Both pack dissolution and the incorporation of non-related wolves into packs would result in decreased local spatial genetic structure, and the differences observed here between pre- and post-harvest suggest that this may be occurring in Minnesota.
In contrast to the local homogenization, population-wide differentiation was higher between the post-harvest genetic clusters compared to pre-harvest, evidenced by division into four subpopulations instead of the three in the year before. The smaller number of migrants post-harvest suggests less gene flow between clusters and less long-distance dispersal between clusters. This is also supported by the decreased number of effective migrants post-harvest (Table 3) and by increased geographic separation between cluster cores after harvest when compared to before harvest, both at the 95 and 99% confidence levels. Such decreased dispersal rates and emigration rates from harvested areas have been observed in wolves in northern Alaska in response to low or moderate harvest (Adams et al. 2008). While this seems to contrast with the hypothesized increase in adopted wolves, there could be increased adoption with reduced gene flow if the adopted individuals are not breeding in their new packs. In our study, decreased gene flow, increased spatial separation, and slightly decreased admixture between clusters in the post-harvest sample year suggest that there were fewer long-distance migrants after the first harvest year. However, the percentage of individuals categorized as admixed increased from 17% in the pre-harvest sample to 27% post-harvest in our study, suggesting that many individuals still have admixed ancestry. Though these numbers suggest increased admixture in the first year post-harvest, the post-harvest number is divided between four clusters instead of three, and thus this increase was expected a priori. It is unknown if this trend will continue in the years following harvest, but these results suggest that harvest corresponds with altered dispersal and gene flow patterns in Minnesota wolves.
There are areas of reduced gene flow in both sample years in the western part of the state, where there are few resident wolf packs, less forested area (more farmland and prairie grasslands), few state- or federally-managed lands, and different prey composition than in northeastern Minnesota. Much of this northwestern area of reduced gene flow covers land broadly classified as agriculture, upland prairie, prairie woodland, or aspen parkland, all of which are more exposed habitat types that may be less favorable wolf habitat than the conifer forest types found in the Laurentian mixed forest farther east (Mladenoff et al. 2009; Treves et al. 2009). An east–west band of reduced gene flow in 2012 corresponds roughly to a highway corridor between Duluth, MN, and Grand Forks, ND. Similarly, both years have a resistive patch just east of Duluth, MN, in a triangular area surrounded by three highways. Though roads themselves do not usually restrict wolf movement across a landscape (Mech et al. 1995; Merrill and Mech 2000), areas with highways tend to have higher human population densities and are less favorable for wolf dispersal (Thiel 1985; Mladenoff et al. 1995). Wolves are highly adaptable and resilient animals and are not necessarily restricted to what has traditionally been considered “favorable” habitat in Minnesota (Mech 2010). However, as wolf densities decrease, there are theoretically more opportunities available for dispersers to disperse to favorable habitat and thus less dispersal into areas with unfavorable habitat, as seems to be the case in the post-harvest sampling year. In other cooperative breeding species, there is evidence for individuals changing dispersal behavior (early vs. delayed dispersal) based on the quality of available territories (Koenig et al. 1992). If most successful wolf pack territories are in favorable habitat, this may result in smaller dispersal distances. However, apparent reductions in dispersal would also result from increased disperser mortality in less favorable areas, and we cannot distinguish between these possibilities in this study.
Increased population genetic structure and a reduction in dispersal or disperser survival may result from recent increases in anthropogenic mortality in the Minnesota wolf population, but further research is necessary to determine if harvest is the primary cause of these differences. Reduced dispersal suggests that more wolves either remained in their natal areas in 2013 or did not successfully disperse. Fewer individuals tend to disperse in demographically stable years compared to years of declining or increasing population in Minnesota (Gese and Mech 1991). The reduced dispersal observed here between pre-harvest and post-harvest samples could presumably result from recent changes in the Minnesota wolf population size, rather than decreased dispersal resulting from harvest pressures. Because population surveys were only conducted every 5 years prior to 2012 (Fig. S1; Erb et al. 2014), we do not know how the population fluctuated between 2007 (the last count prior to harvest) and the beginning of harvest in 2012.
The gene flow patterns observed following harvest contrast with estimates of historical migration rates, which suggest net migration from the interior of wolf range (northeastern Minnesota) toward the perimeter in the state. We additionally found no evidence for a past bottleneck event in the Minnesota wolf population (Supplementary Methods). This supports the findings of Koblmüller et al. (2009) for Great Lakes wolves as a whole, and suggests that the decline and recovery of wolves in Minnesota is more accurately a range contraction and expansion for this widely distributed carnivore, rather than a true population bottleneck across their contiguous range. As the population grew, wolves dispersed toward the edge of occupied range, expanding their range southward and westward.
One major assumption underlying our sampling is that the first year of harvest (2012–2013) was not selective and therefore the harvested wolves represent a random subset of the population. If this assumption is not valid, this could bias our results in the following ways: (a) if dispersers are more at risk of harvest (Murray et al. 2010), then we may detect more gene flow than the actual level of effective migration in the population; (b) if harvest is biased toward dispersers and dispersal behavior is a heritable trait, as Sparkman et al. (2012) suggest for red wolves (Canis rufus), sampling may be biased toward wolves that are more related to one another than the general population; and (c) if the distribution of ages of harvested wolves does not accurately reflect the distribution of ages in the population, then sampling may not be representative of the population as a whole. We do not have evidence for harvest being biased toward a given age or sex of wolf in the years sampled (Stark and Erb 2013, 2014), and no studies have found heritability in dispersal in gray wolves (Mech and Boitani 2003). However, these assumptions and biases should be explored further.
Prey density is also strongly correlated with dispersal in wolf populations (Messier 1985; Ballard et al. 1987) and changes in wolf-to-prey ratios between 2012 and 2014 may have affected gene flow as well. Decreased wolf densities due to hunting pressures increase per-capita prey availability and reduce the dispersal benefit to juveniles (Benson et al. 2014), and thus prey availability is a confounding factor in regulating wolf dispersal. In northern Minnesota, wolves primarily prey on moose (Alces alces) and white-tailed deer (Odocoileus virginianus), with moose more important in northeastern Minnesota and deer more important in the northwestern part of the state (Fritts and Mech 1981; Chenaux-Ibrahim 2015). The moose population across the state has declined in recent years (Delgiudice 2014) and white-tailed deer numbers have also declined, though numbers have stabilized over the past 5–10 years (Grund 2014). The winter of 2013–2014 was especially severe, which impacted prey abundance (D’Angelo and Giudice 2015) and may have affected wolf movement across the state. Further research is needed into climatic and resource conditions between pre-harvest and post-harvest years to determine how these factors may have influenced wolf dispersal, outside of any effect of harvest. Additionally, it is possible that the reduction in gene flow simply results from most dispersers being harvested, rather than altered dispersal behavior, and so investigating patterns in dispersal behavior will illuminate the relative importance of each of these factors.
This study indicates that genetic effects occur on both local and population-wide scales and provides an important foundation for further research into the impact of anthropogenic harvest on the population genetic structure of wolves. Data over a longer time period will be necessary to determine whether the changes observed here continue long-term or if genetic responses change across different time scales. Monitoring the genetic diversity and structure of wild populations will be important in understanding how anthropogenic mortality may positively or negatively affect these populations. In studying population genetic structure, we can determine whether gene flow rates are high enough to maintain connectivity in fragmented populations (Schwartz et al. 2007). The small reductions in gene flow observed in this study do not preclude the possibility of further decreases in gene flow or an increase in immigration from other populations in the long term. In addition, the effects observed in this study appear to be different at different spatial scales: local decreases in population structure are nested within population-wide increases in genetic structure, suggesting that scale is important to detecting these changes. Genetic monitoring has been informative in many harvested species and will be important for appropriately managing harvested wolves worldwide in the future (Schwartz et al. 2007; Creel et al. 2015). Though the Western Great Lakes distinct population segment was re-listed under the ESA in 2014 and currently is federally protected, harvest-based management is likely to resume in the future and this study will be important for long-term population monitoring in this population and others.
References
Adams LG, Stephenson RO, Dale BW et al (2008) Population dynamics and harvest characteristics of wolves in the central Brooks Range, Alaska. Wildl Monogr 170:1–25
Alexander RD (1974) The evolution of social behavior. Annu Rev Ecol Syst 5:325383
Allendorf FW, Hard JJ (2009) Human-induced evolution caused by unnatural selection through harvest of wild animals. Proc Natl Acad Sci 106:9987–9994
Allendorf FW, England PR, Luikart G et al (2008) Genetic effects of harvest on wild animal populations. Trends Ecol Evol 23:327–337
Andreasen AM, Stewart KM, Longland WS et al (2012) Identification of source-sink dynamics in mountain lions of the Great Basin. Mol Ecol 21:5689–5701
Ballard WB, Whitman JS, Gardner CL (1987) Ecology of an exploited wolf population in south-central Alaska. Wildl Monogr 98:3–54
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185:313–326
Benson J, Patterson B, Mahoney P (2014) A protected area influences genotype-specific survival and the structure of a Canis hybrid zone. Ecology 95:254–264
Boitani L (2003) Wolf conservation and recovery. In: Mech LD, Boitani L (eds) Wolves: behavior, ecology, and conservation. The University of Chicago Press, Chicago, pp 317–340
Brainerd SM, Andrén H, Bangs EE et al (2008) The effects of breeder loss on wolves. J Wildl Manag 72:89–98
Breen M, Jouquand S, Renier C et al (2001) Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res 11:1784–1795
Caniglia R, Fabbri E, Galaverni M et al (2014) Noninvasive sampling and genetic variability, pack structure, and dynamics in an expanding wolf population. J Mammal 95:41–59
Chenaux-Ibrahim Y (2015) Seasonal diet composition of gray wolves (Canis lupus) in northeastern Minnesota determined by scat analysis. Thesis, University of Minnesota
Coltman DW (2008) Molecular ecological approaches to studying the evolutionary impact of selective harvesting in wildlife. Mol Ecol 17:221–235
Coltman DW, O’Donoghue P, Jorgenson JT et al (2003) Undesirable evolutionary consequences of trophy hunting. Nature 426:655–658
Creel S, Becker M, Christianson D et al (2015) Questionable policy for large carnivore hunting. Science 350:1473–1475
D’Angelo GJ, Giudice JH (2015) Monitoring population trends of white-tailed deer in Minnesota. Minnesota Department of Natural Resources, St. Paul. http://files.dnr.state.mn.us/wildlife/deer/reports/harvest/popmodel_2015.pdf
Delgiudice GD (2014) 2014 Aerial Moose Survey. Minnesota Department of Natural Resources, St. Paul, MN. 6p. http://files.dnr.state.mn.us/wildlife/moose/2014_moosesurvey.pdf
Do C, Waples RS, Peel D et al. (2014) NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Erb J, Sampson B (2013) Distribution and abundance of wolves in Minnesota, 2012–2013. Minnesota Department of Natural Resources, Grand Rapids. http://files.dnr.state.mn.us/fish_wildlife/wildlife/wolves/2013/wolfsurvey_2013.pdf
Erb J, Humpal C, Sampson B (2014) Minnesota wolf population update 2014. Minnesota Department of Natural Resources, Grand Rapids, 7p. http://files.dnr.state.mn.us/wildlife/wolves/2014/survey_wolf.pdf
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fenberg FB, Roy K (2008) Ecological and evolutionary consequences of size-selective harvesting: how much do we know? Mol Ecol 17:209–220
Francisco LV, Langston AA, Mellersh CS et al (1996) A class of highly polymorphic tetranucleotide repeats for canine genetic mapping. Mamm Genome 7:359–362
Frank SA (1986) Dispersal polymorphism in subdivided populations. J Theor Biol 122:303–309
Frankel OH, Soulé ME (1981) Conservation and evolution. Cambridge University Press, Cambridge
Frankham R (2005) Genetics and extinction. Biol Conserv 126:131–140
Frantz AC, Cellina S, Krier A et al (2009) Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: Clusters or isolation by distance? J Appl Ecol 46:493–505
Fredholm M, Winterø AK (1996) Efficient resolution of parentage in dogs by amplification of microsatellites. Anim Genet 27:19–23
Fritts SH (1983) Record dispersal by a wolf from Minnesota. J Mammal 64:166–167
Fritts SH, Mech LD (1981) Dynamics, movements, and feeding ecology of a newly protected wolf population in northwestern Minnesota. Wildl Monogr 80:3–79
Fritts SH, Stephenson RO, Hayes RD, Boitani L (2003) Wolves and humans. In: Mech LD, Boitani L (eds) Wolves: behavior, ecology, and conservation. The University of Chicago Press, Chicago, pp 289–316
Fuller TK (1989) Population dynamics of wolves in north-central Minnesota. Wildl Monogr 105:3–41
Fuller TK, Berg WE, Radde GL et al (1992) A history and current estimate of wolf distribution and numbers in Minnesota. Wildl Soc Bull 20:42–55
Garant D, Forde SE, Hedry AP (2007) The multifarious effects of dispersal and gene flow on contemporary adaptation. Funct Ecol 21:434–443
Gese EM, Mech LD (1991) Dispersal of wolves (Canis lupus) in northeastern Minnesota, 1969–1989. Can J Zool 69:2946–2955
Getis A, Ord JK (1992) The analysis of spatial association. Geogr Anal 24:189–206
Goudet J (1995) FSTAT (version 1.2): A computer program to calculate F-statistics. J Hered 86:485–486
Greenwood PJ, Harvey PH, Perrins CM (1978) Inbreeding and dispersal in the great tit. Nature 271:52–54
Grewal SK, Wilson PJ, Kung TK et al (2004) A genetic assessment of the Eastern wolf (Canis lycaon) in Algonquin Provincial Park. J Mammal 85:625–632
Grund M (2014) Monitoring population trends of white-tailed deer in Minnesota. Minnesota Department of Natural Resources, St. Paul.http://files.dnr.state.mn.us/wildlife/deer/reports/harvest/popmodel_2014.pdf
Guyon R, Lorentzen TD, Hitte C et al (2003) A 1-Mb resolution radiation hybrid map of the canine genome. Proc Natl Acad Sci 100:5296–5301
Hamilton WD, May RM (1977) Dispersal in stable habitats. Nature 269:578–581
Hard JJ, Gross MR, Heino M et al (2008) Evolutionary consequences of fishing and their implications for salmon. Evol Appl 1:388–408
Harris RB, Wall WA, Allendorf FW (2002) Genetic consequences of hunting: what do we know and what should we do? Wildl Soc Bull 30:634–643
Jakobsson M, Rosenberg NA (2007) CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jędrzejewski W, Branicki W, Veit C et al (2005) Genetic diversity and relatedness within packs in an intensely hunted population of wolves Canis lupus. Acta Theriol 50:3–22
Jennions MD, Macdonald DW (1994) Cooperative breeding in mammals. Trends Ecol Evol 9:89–93
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Kimura M, Weiss G (1964) The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49:561–576
Koblmüller S, Nord M, Wayne RK, Leonard JA (2009) Origin and status of the Great Lakes wolf. Mol Ecol 18:2313–2326
Koenig WD, Pitelka FA, Carmen WJ et al (1992) The evolution of delayed dispersal in cooperative breeders. Q Rev Biol 67:111–150
Koenig WD, Dickinson JL, Emlen ST (2016) Synthesis: Cooperative breeding in the twenty-first century. In: Koenig WD, Dickinson JL (eds) Cooperative breeding in vertebrates: studies of ecology, evolution, and behavior. Cambridge University Press, Cambridge, pp 353–374
Kuparinen A, Merilä (2007) Detecting and managing fisheries-induced evolution. Trends Ecol Evol 22:652–659
Lehman N, Clarkson P, Mech LD et al (1992) A study of the genetic relationships within and among wolf packs using DNA fingerprinting and mitochondrial DNA. Behav Ecol Sociobiol 30:83–94
McCullough DR (1996) Spatially structured populations and harvest theory. J Wildl Manag 60:1–9
Mech LD (1999) Alpha status, dominance, and division of labor in wolf packs. Can J Zool 77:1196–1203
Mech LD (2006) Estimated age structure of wolves in Northeastern Minnesota. J Wildl Manag 70:1481–1483
Mech LD (2010) Prediction failure of a wolf landscape model. Wildlife Soc B 34:874–877
Mech LD, Boitani L (2003) Wolf Social Ecology. In: Mech LD, Boitani L (eds) Wolves: behavior, ecology, and conservation. The University of Chicago Press, Chicago pp 1–34
Mech LD, Fritts SH, Wagner D (1995) Minnesota wolf dispersal to Wisconsin and Michigan. Am Midl Nat 133:368–370
Merrill S, Mech LD (2000) Details of extensive movements by Minnesota wolves (Canis lupus). Am Midl Nat 144:428–433
Messier F (1985) Social organization, spatial distribution and population density of wolves in relation to moose density. Can J Zool 63:1068–1077
Meyers-Wallen VN, Palmer VL, Acland GM, Hershfield B (1995) Sry-negative XX sex reversal in the American cocker spaniel dog. Mol Reprod Dev 41:300–305
Milleret C, Wabakken P, Liberg O et al (2016) Let’s stay together? Intrinsic and extrinsic factors involved in pair bond dissolution in a recolonizing wolf population. J Anim Ecol. doi:10.1111/1365-2656.12587
Milner JM, Nilsen JM, Andreassen HP (2007) Demographic side effects of selective hunting in ungulates and carnivores. Conserv Biol 21:36–47
Mladenoff DJ, Sickley TA, Haight RG, Wydeven AP (1995) A regional landscape analysis and prediction of favorable gray wolf habitat in the northern Great Lakes region. Conserv Biol 9:279–294
Mladenoff DJ, Clayton MK, Pratt SD et al (2009) Change in occupied wolf habitat in the northern Great Lakes region. In: Wydeven AP, Van Deelen TR, Heske EJ (eds) Recovery of gray wolves in the Great Lakes region of the United States. Springer Science, New York, pp 119–138
Murray DL, Smith DW, Bangs EE et al (2010) Death from anthropogenic causes is partially compensatory in recovering wolf populations. Biol Conserv 143:2514–2524
Neff MW, Broman KW, Mellersh CS et al (1999) A second-generation genetic linkage map of the domestic dog, Canis familiaris. Genetics 151:803–820
Newby JR, Scott Mills L, Ruth TK et al (2013) Human-caused mortality influences spatial population dynamics: Pumas in landscapes with varying mortality risks. Biol Conserv 159:230–239
Olivieri I, Michalakis Y, Gouyon PH (1995) Metapopulation genetics and the evolution of dispersal. Am Nat 146:202–228
Ostrander EA, Sprague G, Rine J (1993) Identification and characterization of dinucleotide repeat (CA)n markers for genetic mapping in dog. Genomics 16:207–213
Peakall R, Smouse PE (2012) GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539
Petkova D, Novembre J, Stephens M (2014) Visualizing spatial population structure with estimated effective migration surfaces. bioRxiv doi:10.1101/011809
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Rich LN, Mitchell MS, Gude JA, Sime CA (2012) Anthropogenic mortality, intraspecific competition, and prey availability influence territory sizes of wolves in Montana. J Mammal 93:722–731
Richman M, Mellersh CS, André C et al (2001) Characterization of a minimal screening set of 172 microsatellite markers for genome-wide screens of the canine genome. J Biochem Biophys Methods 47:137–149
Ronce O (2007) How does it feel to be like a rolling stone? Ten questions about dispersal evolution. Annu Rev Ecol Evol Syst 38:231–253
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106.
Rutledge LY, Patterson BR, Mills KJ et al (2010) Protection from harvesting restores the natural social structure of eastern wolf packs. Biol Conserv 143:332–339
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Schwartz MK, McKelvey KS (2009) Why sampling scheme matters: The effect of sampling scheme on landscape genetic results. Conserv Genet 10:441–452
Schwartz MK, Luikart G, Waples RS (2007) Genetic monitoring as a promising tool for conservation and management. Trends Ecol Evol 22:25–33
Smouse PE, Peakall R (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82:561–573
Sparkman AM, Waits LP, Murray DL (2011) Social and demographic effects of anthropogenic mortality: a test of the compensatory mortality hypothesis in the red wolf. PLoS ONE 6:e20868
Sparkman AM, Adams JR, Steury TD et al (2012) Evidence for a genetic basis for delayed dispersal in a cooperatively breeding canid. Anim Behav 83:1091–1098
Stark D, Erb J (2013) 2012 Minnesota wolf season report. Minnesota Department of Natural Resources, Grand Rapids. http://files.dnr.state.mn.us/fish_wildlife/wildlife/wolves/2013/wolfseasoninfo_2012.pdf
Stark D, Erb J (2014) 2013 Minnesota wolf season report. Minnesota Department of Natural Resources, Grand Rapids. http://files.dnr.state.mn.us/recreation/hunting/wolf/2013-wolf-season-report.pdf
Thiel RP (1985) The relationships between road densities and wolf habitat in Wisconsin. Am Midl Nat 113:404–407
Treves A, Martin KA, Wiedenhoeft JE, Wydeven AP (2009) Dispersal of gray wolves in the Great Lakes region. In: Wydeven AP, Van Deelen TR, Heske EJ (eds) Recovery of gray wolves in the Great Lakes region of the United States. Springer Science, New York, pp 191–204
U.S. Fish and Wildlife Service (2014) Western Great Lakes Distinct Population Segment of the Gray Wolf 2012–2014 Post Delisting Monitoring Annual Report. U.S. Fish and Wildlife Service, Twin Cities Field Office and Midwest Region, Bloomington. https://www.fws.gov/midwest/wolf/monitoring/pdf/Year1PDMReportSept2014.pdf
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Waser PM, Austad SN, Keane B (1986) When should animals tolerate inbreeding? Am Nat 128:529–537
Wayne RK, Vilà C (2003) Molecular genetic studies of wolves. In: Mech LD, Boitani L (eds) Wolves: behavior, ecology, and conservation. University of Chicago Press, Chicago, pp 218–238
Webb N, Allen J, Merrill E (2011) Demography of a harvested population of wolves (Canis lupus) in west-central Alberta, Canada. Can J Zool 752:744–752
Acknowledgements
The authors would like to thank the Minnesota Department of Natural Resources for providing the samples used in this study. We would also like to thank D. Petkova and J. Novembre for discussing the use and interpretation of EEMS. Earlier versions of this manuscript were greatly improved by comments from B. Gross, J. Pastor, C. Wagner, and J. Alston. RAM was partially supported by funding from the Minnesota State Environmental and Natural Resources Trust Fund. JLS and JAR were supported by funding from the University of Minnesota- Duluth.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rick, J.A., Moen, R.A., Erb, J.D. et al. Population structure and gene flow in a newly harvested gray wolf (Canis lupus) population. Conserv Genet 18, 1091–1104 (2017). https://doi.org/10.1007/s10592-017-0961-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-017-0961-7