
Abstract
Major histocompatibility complex (MHC) genes are excellent markers for the study of adaptive genetic variation occurring over different geographical scales. The Chinese egret (Egretta eulophotes) is a vulnerable ardeid species with an estimated global population of 2600–3400 individuals. In this study, we sampled 172 individuals of this egret (approximately 6 % of the global population) from five natural populations that span the entire distribution range of this species in China. We examined their population genetic diversity and geographical differentiation at three MHC class II DAB genes by identifying eight exon 2 alleles at Egeu-DAB1, eight at Egeu-DAB2 and four at Egeu-DAB3. Allelic distributions at each of these three Egeu-DAB loci varied substantially within the five populations, while levels of genetic diversity varied slightly among the populations. Analysis of molecular variance showed low but significant genetic differentiation among five populations at all three Egeu-DAB loci (haplotype-based ϕST: 0.029, 0.020 and 0.042; and distance-based ϕST: 0.036, 0.027 and 0.043, respectively; all P < 0.01). The Mantel test suggested that this significant population genetic differentiation was likely due to an isolation-by-distance pattern of MHC evolution. However, the phylogenetic analyses and the Bayesian clustering analysis based on the three Egeu-DAB loci indicated that there was little geographical structuring of the genetic differentiation among five populations. These results provide fundamental population information for the conservation genetics of the vulnerable Chinese egret.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The generation and maintenance of genetic variation within or among natural populations is a central issue in evolutionary and conservation biology (Piertney and Oliver 2006). Information on adaptive genetic variation at different geographical scales can be efficiently used to inform conservation management such as identifying adaptive units (AUs, special conservation units which are based on patterns of adaptive differentiation) (Funk et al. 2012; Zhu et al. 2013). Typically, studies of genetic variation occurring over different geographical scales have used neutral or nearly neutral markers, such as mitochondrial DNA (mtDNA), microsatellites or SNPs (Martínez-Cruz et al. 2004; Morin et al. 2004; Hull et al. 2008; Addis et al. 2015; Corrêa et al. 2015). Although these neutral markers can be used to infer demographic events or population history in natural populations (Fabiani et al. 2003; Ekblom et al. 2007; Zhou et al. 2010; Corrêa et al. 2015), their ability to demonstrate adaptive variation is limited (Meyers and Bull 2002; Alcaide et al. 2008; Witzenberger and Hochkirch 2011). Generally, neutral markers provide demographic history while adaptive markers show how these populations adapt to the environment (Ekblom et al. 2007; Alcaide et al. 2008; Vásquez-Carrillo et al. 2013). Neutral genetic variation is influenced by demographic factors, such as genetic drift and gene flow, whereas adaptive genetic variation is affected by both demographic factors and selective factors (Bichet et al. 2015; Lillie et al. 2015). Therefore, adaptive genetic markers can be efficiently used to study population adaptive variation at different environments. Recently, Major histocompatibility complex (MHC) genes have been used to study adaptive variation at different geographical scales, because the variation in pathogens across regions may lead to differential selection pressure on MHC proteins (Sommer 2005; Piertney and Oliver 2006; Spurgin and Richardson 2010).
The MHC is a multi-gene family that plays important roles in susceptibility or resistance to many vertebrate diseases, principally by recognizing foreign peptides and presenting them to T cells, initiating the adaptive immune response (Klein 1986; Klein et al. 1993; Sommer 2005; Janeway et al. 2008). Traditionally, MHC genes are classified into two major classes: class I and class II, which typically present peptide antigens that arise from intracellular and extracellular proteins, to CD8+ and CD4+ T cells, respectively (Bevan 1987; Germain et al. 1996; Janeway et al. 2008). MHC class II genes encode heterodimers composed of alpha- and beta-chains (Klareskog et al. 1977), and thus can be further subdivided into A (e.g., DRA) and B (e.g., DAB, DQB and DPB) genes, in which the B genes are responsible for the majority of the polymorphism. Which foreign peptides an individual can respond to is largely determined by genetic variation at specific regions of MHC genes (exons 2 and 3 of class I, and exon 2 of class II), which in turn influence individual fitness and long-term survival of populations (Klein 1986; Hughes 1991; Hughes and Nei 1992; Janeway et al. 2008). Genetic variation of the MHC is postulated to be generated by gene duplication and deletion, intra- and inter-locus recombination or gene conversion, and the accumulation of de novo mutations (Nei and Rooney 2005; Balakrishnan et al. 2010; Spurgin et al. 2011; Promerová et al. 2013; Nguyen-Phuc et al. 2016). MHC polymorphism is maintained by some forms of balancing selection, such as frequency-dependent selection (Takahata and Nei 1990), heterozygous advantage (Doherty and Zinkernagel 1975) and MHC-dependent mate choice (Penn and Potts 1999).
Recently, studies of geographical variation at MHC loci have been reported for several groups of Aves, including Passeriformes (Miller and Lambert 2004; Aguilar et al. 2005; Schut et al. 2011; Jones et al. 2014; Bichet et al. 2015), Galliformes (Piertney 2003; Nguyen-Phuc et al. 2016; Zeng et al. 2016), Charadriiformes (Ekblom et al. 2007; Vásquez-Carrillo et al. 2013), Strigiformes (Kohyama et al. 2015), and Falconiformes (Alcaide et al. 2008). However, as far as we know, there has hitherto been no study of MHC geographical variation conducted in Ciconiiformes, particularly in threatened ardeid birds.
The Chinese egret (Ciconiiformes, Ardeidae, Egretta eulophotes) is a migratory colonial waterbird, wintering in the south of Asia while breeding on offshore islands in Russia, North Korea, South Korea and China. This egret was nominated by Swinhoe after he collected the type specimen from Xiamen (formerly known as Amoy), China in 1863. Its populations have been declining dramatically since the nineteenth century (Kushlan and Hancock 2005; BirdLife International 2015; IUCN 2015). Currently, this egret is listed as a vulnerable species with an estimated global population of 2600–3400 individuals (BirdLife International 2015; IUCN 2015). In our previous MHC studies on this vulnerable species, we have isolated and characterized three classical single-copy loci of the MHC class II DAB gene (named as Egeu-DAB1, -DAB2, and -DAB3), and established an efficient locus-specific MHC genotyping technique (Li et al. 2011; Wang et al. 2013; Lei et al. 2015). Furthermore, our previous study (Zhou et al. 2010) found that there was a relatively high level of mtDNA genetic diversity in three populations of Chinese egret in China, and these populations had low but significant genetic differentiation with little geographical structure. To expand upon our previous work, providing the first study of population genetic diversity and differentiation at the MHC in ciconiiform birds, we had the following specific aims: (1) to analyze the allelic distribution at exon 2 of three Egeu-DAB genes in five populations of the Chinese egret in China; (2) to examine the genetic diversity of these genes in these five populations; and (3) to analyze the genetic differentiation of Egeu-DAB genes among these five populations that span the entire distribution range of this species in China. Our results provide fundamental population information for the conservation genetics of the vulnerable Chinese egret.
Materials and methods
Sample collection and DNA extraction
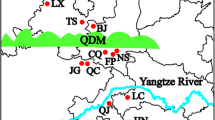
Sample collection from Chinese egret was conducted during the morning (06:00–08:00), and visits to the breeding colonies were restricted to a maximum of 2 h every day. A total of 172 feather samples of nestlings, originating from 172 nests, were individually collected from five archipelago populations: Xingrentuo (Xrt; 39°31′N, 123°03′E; n = 38), Hailvdao (Hld; 37°26′N, 122°40′E; n = 28), Mantoushan (Mts; 30°13′N, 121°53′E; n = 35), Riyu (Ry; 27°01′N, 120°25′E; n = 34) and Xiaocaiyu (Xcy; 23°48′N, 117°45′E; n = 37). The locations of these sampling archipelagoes span the entire Chinese distribution range of the Chinese egret, and can be taken to represent the different geographical populations across China (Fig. 1). The feather samples were preserved in 95 % ethanol and frozen at −80 °C. Genomic DNA was extracted using the Universal Genomic DNA Extraction Kit Ver. 3.0 (Takara, Dalian, China) following the manufacturer’s protocols, and then was kept at −80 °C until further use.

Geographical locations of the sampled Chinese egret populations in China. Xrt Xingrentuo, Hld Hailvdao, Mts Mantoushan, Ry Riyu, Xcy Xiaocaiyu
PCR and SSCP genotyping
Genetic polymorphism of exon 2 sequences at the three single Egeu-DAB loci was examined by semi-nested asymmetric polymerase chain reaction (PCR) combined with single-strand conformation polymorphism (SSCP), as previously described (Lei et al. 2015). Briefly, to first produce single-stranded amplicons, three-round PCRs, including the asymmetric PCR, were carried out. The single-stranded amplicons were then loaded on 10 % non-denaturing polyacrylamide gels (PAGEs) for electrophoresis, and visualized by the sensitive silver-staining procedure. Finally, SSCP-bands were excised from gels, re-amplified and sequenced following the protocols of Wang et al. (2013). To avoid the inclusion of PCR artifacts, every allele was directly sequenced in both directions from at least two individuals, or from two independent PCRs from one individual. Throughout this study, the word “allele” is used to describe the full-length exon 2 sequence (270 bp), of all three Egeu-DAB loci, derived from SSCP genotyping.
Data analyses
Exon 2 sequences obtained from the 172 individuals were aligned and edited using BioEdit v7.0.5.3 (Hall 1999). Estimates of allele frequency, observed heterozygosity and expected heterozygosity, and tests of deviation from Hardy–Weinberg equilibrium were assessed using GENEPOP 4.0 (Rousset 2008). Calculations of gene diversity and nucleotide diversity were conducted in FSTAT 1.2 (Goudet 1995) and DnaSP v5 (Librado and Rozas 2009), respectively. The pairwise nucleotide distance (p-distance) among haplotypes was calculated using MEGA 6.0 (Tamura et al. 2013). Allelic richness, a measure of the number of alleles independent of sample size, was estimated with FSTAT 1.2 (Goudet 1995).
Calculations of fixation index (ϕST) and pairwise comparison FST, analysis of molecular variance (AMOVA) and Mantel test were carried out with Arlequin 3.5 (Excoffier and Lischer 2010). ϕST, pairwise FST and AMOVA were based on both the haplotype frequencies and the molecular distances among haplotypes, using the Kimura two-parameter model. Statistical significance of the observed variance was determined using 1023 haplotype permutations. A Mantel test was carried out with 10,000 permutations, to investigate the isolation-by-distance relationship between the estimate of FST/(1 − FST) and the natural logarithm of geographic distance. Geographical distance (in km) was measured using Google Earth (http://earth.google.com), based on a straight line connecting each pair of sampled populations.
Phylogenetic relationships of exon 2 nucleotide sequences were analyzed separately for each of the three Egeu-DAB loci. PartitionFinder v1.1.1 (Lanfear et al. 2012) was used to determine the best-fit nucleotide substitution model, according to the Bayesian information criterion (BIC) and a “greedy” algorithm with branch lengths estimated as “unlinked”. The analyses suggested that the proposed best-fit nucleotide substitution model for each of the three locus-specific datasets was Kimura two-parameter model with gamma distribution substitution rates and proportion of invariable sites (BIC score: 1600.76, 1454.87, and 1258.68, respectively). These results were then implemented in phylogenetic analyses, which were conducted using the maximum likelihood method with 1000 bootstrap replicates in MEGA 6.0 (Tamura et al. 2013).
The Bayesian population clustering program STRUCTURE 2.3.3 (Falush et al. 2003) was used to investigate differentiation across the five Chinese egret populations. The structure analysis was conducted using an admixture model with correlated allele frequencies, and was run from K = 1 to 10 with 10 runs per K and a burn-in of 100,000 and 1,000,000 reps after the burn-in. The results were then uploaded to the Structure Harvester server (http://taylor0.biology.ucla.edu/structureHarvester/), which selects the number of clusters by simultaneously evaluating posterior probability and the Delta K statistic of Evanno et al. (2005).
Results
Allelic distribution within different populations
In the 172 examined individuals from five Chinese egret populations, a total of eight, eight and four exon 2 alleles were identified at Egeu-DAB1, -DAB2 and -DAB3 loci, respectively (Tables 1, S1, Supplementary material). For each locus, no stop codons or insertions or deletions were observed, and typically 1–2 alleles were identified per individual, suggesting that for each locus only one gene copy was sequenced with the primer sets. Sequences of these confirmed alleles were submitted to GenBank, with names according to the nomenclature proposed by Klein et al. (1990), denoted by the species’ gene prefix (Egeu-DAB), with a suffix comprising a locus number (1–3) and two sequential allele numbers (01–12). Their accession numbers are listed in Table 1.
The deviation from Hardy–Weinberg equilibrium within each locus in each population was statistically significant (all P < 0.05). Allelic distributions of the three Egeu-DAB loci varied substantially within the five Chinese egret populations (Table 1). Egeu-DAB1*12 was detected in only one population (Hld), while the remaining 19 alleles were shared between at least two populations. The most frequent alleles shared were Egeu-DAB1*01, *02, *03, *06, Egeu-DAB2*01, *02, *04, and Egeu-DAB3*01, *02, which were found in all populations, at different frequencies. No single Egeu-DAB1 allele was most common in all populations. Egeu-DAB1*01 was most common in Hld (allele frequency: 0.357) and Xcy (0.351), while Egeu-DAB1*03 was most common in Xrt (0.329), Mts (0.257) and Ry (0.353). However, a single Egeu-DAB2 allele (Egeu-DAB2*01) and a single Egeu-DAB3 allele (Egeu-DAB3*01) was most common across all five studied populations, with frequencies higher than 0.45 (Table 1). More specifically, Egeu-DAB3*01 was highly abundant (allele frequency: >0.75) in all populations, and its frequency (0.776 → 0.786 → 0.871 → 0.912 → 0.959) increased with decreasing latitude (39°31′ → 37°26′ → 30°13′ → 27°01′ → 23°48′).
Genetic diversity in different populations
Several genetic diversity parameters were calculated to estimate genetic diversity of the vulnerable Chinese egret, including gene diversity (Gd), nucleotide diversity (π), allelic richness (AR) and expected heterozygosity (He). Detailed diversity statistics of the three Egeu-DAB loci within the five populations are summarized in Table 2. At each Egeu-DAB locus, the total number of haplotypes (N), Gd, π, AR, observed heterozygosity (Ho) and He were all found to vary slightly among the five populations. The mean AR across populations was 6.192 ± 0.202 for Egeu-DAB1, 6.022 ± 0.531 for Egeu-DAB2 and 2.734 ± 0.192 for Egeu-DAB3. Relatively high level of genetic diversity was found at the Egeu-DAB1 locus, as indicated by high values of Gd (0.744–0.812), π (0.056–0.067), AR (5.984–6.999) and He (0.737–0.806) (Table 2). Estimation of heterozygosity revealed significantly (P < 0.05) lower levels of observed heterozygosity than expected for all five populations at Egeu-DAB1; for Hld and Ry at Egeu-DAB2; and for Xrt, Mts and Ry at Egeu-DAB3. The average pairwise nucleotide distances among the haplotypes of the three Egeu-DAB genes were 0.077 ± 0.005, 0.061 ± 0.005 and 0.083 ± 0.008, respectively.
Genetic differentiation among different populations
The AMOVA revealed low but highly significant ϕST values for the Egeu-DAB1 locus, whether based on haplotype frequencies (0.029, P < 0.01) or molecular distances (0.036, P < 0.01), with the majority of variance being found within populations (haplotype-based: 97.11 % and distance-based: 96.40 %) rather than among populations (2.89 and 3.60 %) (Table 3). Similar AMOVA results were shown for Egeu-DAB2, -DAB3, and all three loci combined (Egeu-DAB), suggesting that low but significant genetic differentiation was present among the five populations (haplotype-based ϕST: 0.020 for -DAB2, 0.042 for -DAB3, 0.007 for -DAB; and distance-based ϕST: 0.027, 0.043, 0.005, respectively; all P < 0.05) (Table 3).
To further assess the genetic differentiation between populations, the pairwise comparison FST values were calculated (Table 4). Pairwise FST values based on haplotype frequencies estimated ranged from −0.016 to 0.097, while those based on molecular distances estimated ranged from −0.016 to 0.110, for the three Egeu-DAB loci. Forty-two out of the 60 (70.00 %) FST values were lower than 0.05 (Table 4), indicating low genetic differentiation among populations.
The Mantel test indicated that there was a significant isolation-by-distance pattern at the Egeu-DAB1 locus, comparing the FST/(1 − FST) value based on estimated molecular distances, with the natural logarithm of the geographic distance (r = 0.410, P = 0.047), however no significant isolation-by-distance relationship was suggested when FST/(1 − FST) was based on estimated haplotype frequencies (r = 0.275, P = 0.132) (Fig. 2a). No significant isolation-by-distance pattern at the Egeu-DAB2 locus was suggested, either based on estimated haplotype frequencies (r = 0.462, P = 0.129) or estimated molecular distances (r = 0.420, P = 0.146) (Fig. 2b). Significant evidence for an isolation-by-distance pattern was detected at the Egeu-DAB3 locus (haplotype-based: r = 0.903, P = 0.009 and distance-based: r = 0.893, P = 0.008) (Fig. 2c).

Isolation-by-distance, with pairwise comparisons of the five Chinese egret populations. a Comparisons at the Egeu-DAB1 locus; b Comparisons at the Egeu-DAB2 locus; c Comparisons at the Egeu-DAB3 locus. Filled triangles represent pairwise comparison values based on estimated haplotype frequencies, while open triangles represent values based on estimated molecular distances
Maximum likelihood trees of each of the three Egeu-DAB exon 2 nucleotide sequences showed little internal resolution, with sequences not grouped according to sampling location. This suggests that our phylogenetic analysis of the sequences does not demonstrate geographical structure of the genetic differentiation of the Chinese egret (Fig. 3). This absence of geographical structure was also suggested by the Bayesian clustering analysis, based on the three Egeu-DAB loci (Fig. 4). Although the Delta K showed one peak at K = 2 (Fig. S1, Supplementary material), the STRUCTURE analysis indicated very weak subdivision, where most egrets showed high levels of admixture between the two putative genetic clusters.

Maximum likelihood trees showing the phylogenetic relationships between Egeu-DAB alleles among the five Chinese egret populations. a Egeu-DAB1 alleles; b Egeu-DAB2 alleles; c Egeu-DAB3 alleles. Each allele is first denoted by the species’ gene prefix (Egeu), followed by a locus number (1–3) and two sequential allele numbers (01–12). Bootstrap values greater than 50 %, and the names of sampling locations (Xrt Xingrentuo, Hld Hailvdao, Mts Mantoushan, Ry Riyu, Xcy Xiaocaiyu) are shown

Genetic structure of the five Chinese egret populations in China. The genetic structure is based on the three Egeu-DAB loci and inferred by Bayesian clustering analysis, with the sampling location as prior information. Each bar represents the probability (P, y-axis) that an individual belongs to a particular color-coded population
Discussion
In this study, many Egeu-DAB alleles were found to be shared among populations, while only one population-specific allele was detected (Egeu-DAB1*12 in Hld, Table 1) suggesting a recent common historical past of these populations (Corrêa et al. 2015). On the other hand, our previous study found significant signs of positive selection in all the three Egeu-DAB loci, with dN/dS (dN, rate of non-synonymous substitution; dS, rate of synonymous substitution) ratios of the putative peptide-binding region being significantly greater than one (Lei et al. 2016). These findings indicated that most MHC alleles of the Chinese egret might have been conserved by positive selection (Weber et al. 2004; Luo et al. 2012), while Egeu-DAB1*12 might be the consequence of adaptive evolution occurred in Hld (e.g., responding to new pathogen variant). In addition, common alleles differed in frequency between populations. These common alleles might represent ancient sequences that have been selectively maintained in populations (Koutsogiannouli et al. 2014). The frequency of one of these common alleles, Egeu-DAB3*01, increased from north to south, which may correlate with the intensity of selective pressure from pathogens to which Egeu-DAB3*01 could respond. To assure the reliability of this conclusion, pathogen community characterization of the Chinese egret is needed.
Among the five Chinese egret populations, there was low but significant genetic differentiation, with little geographical structure, based on the following findings: (1) the AMOVA revealed low but highly significant ФST values for all three Egeu-DAB loci, using either haplotype frequencies or molecular distances; (2) the pairwise population analysis returned most FST values lower than 0.05, indicating low genetic differentiation among populations; (3) the phylogenetic analyses showed that there was no obvious geographical separation of Egeu-DAB alleles in any of the Maximum likelihood trees; (4) the Bayesian clustering analysis indicated very weak population subdivision at the three MHC loci. These MHC-based results are in accord with our previous mitochondrial data in the Chinese egret, which showed that three (Xrt, Ry, and Xcy) of the five populations sampled in this study had low but significant genetic differentiation with little geographical structure (Zhou et al. 2010). The Mantel test further suggested that the low but significant MHC genetic differentiation among our studied populations was likely due to an isolation-by-distance pattern, a commonly observed phenomenon in natural populations. It is defined as a decrease in the genetic similarity among populations as the geographic distance between them increases (Slatkin 1993; Jensen et al. 2005; Alcaide et al. 2008).
Population genetics data collected from adaptive loci, such as the MHC, can provide significant information on adaptive variation, making them useful for identifying AUs in threatened species (Funk et al. 2012; Zhu et al. 2013). Ideally, combining information from neutral and adaptive genetic markers can provide a clearer delineation of conservation units (Vásquez-Carrillo et al. 2013). The results of this new MHC approach complemented with the previous study of mtDNA neutral loci (Zhou et al. 2010) are unanimous in support that all Chinese egret populations in China might be considered as a single AU for conservation management. Although the Bayesian clustering analysis indicated that there were two possible genetic clusters, the five populations can be considered as a single AU based on our data, because most egrets in these two clusters showed high levels of admixture. This admixture could be related to the migratory colonial life pattern of the Chinese egret wintering in the south of Asia while breeding in Russia, North Korea, South Korea, and China (Kushlan and Hancock 2005; Zhou et al. 2010; IUCN 2015). Recently, it has been suggested that diversifying selection acts in a non-uniform manner across the entire MHC region (Smith et al. 2011), and pathogen data within wild populations would directly reflect the different characteristics of possible adaptive groups (Zhu et al. 2013). Nevertheless, MHC loci are unlikely to represent overall adaptive variation, and immune molecules derived from both the innate and adaptive immune system are significant in mounting an immune response to pathogens (Acevedo-Whitehouse and Cunningham 2006; Poelstra et al. 2014). Therefore, in future studies, it will be necessary to further clarify the genetic variation of Chinese egret populations using additional innate or adaptive immune system loci (e.g., Toll-like receptor, TLR; MHC class I UAA, class II DRA and DQB) combined with population pathogen data (e.g., parasite load), as well as using more comprehensive population sampling from a worldwide geographical range.
References
Acevedo-Whitehouse K, Cunningham AA (2006) Is MHC enough for understanding wildlife immunogenetics? Trends Ecol Evol 21:433–438. doi:10.1016/j.tree.2006.05.010
Addis BR, Lowe WH, Hossack BR, Allendorf FW (2015) Population genetic structure and disease in montane boreal toads: more heterozygous individuals are more likely to be infected with amphibian chytrid. Conserv Genet 16:833–844. doi:10.1007/s10592-015-0704-6
Aguilar A, Smith TB, Wayne RK (2005) A comparison of variation between a MHC pseudogene and microsatellite loci of the little greenbul (Andropadus virens). BMC Evol Biol 5:47. doi:10.1186/1471-2148-5-47
Alcaide M, Edwards SV, Negro JJ, Serrano D, Tella JL (2008) Extensive polymorphism and geographical variation at a positively selected MHC class II B gene of the lesser kestrel (Falco naumanni). Mol Ecol 17:2652–2665. doi:10.1111/j.1365-294X.2008.03791.x
Balakrishnan CN, Ekblom R, Völker M, Westerdahl H, Godinez R, Kotkiewicz H, Burt DW, Graves T, Griffin DK, Warren WC, Edwards SV (2010) Gene duplication and fragmentation in the zebra finch major histocompatibility complex. BMC Biol 8:29. doi:10.1186/1741-7007-8-29
Bevan MJ (1987) Class discrimination in the world of immunology. Nature 325:192–194. doi:10.1038/325192b0
Bichet C, Moodley Y, Penn DJ, Sorci G, Garnier S (2015) Genetic structure in insular and mainland populations of house sparrows (Passer domesticus) and their hemosporidian parasites. Ecol Evol 5:1639–1652. doi:10.1002/ece3.1452
BirdLife International (2015) Species factsheet: Egretta eulophotes. Available via DIALOG. http://www.birdlife.org. Accessed 27 Jan 2015
Corrêa TC, Del Lama SN, De Souza JR, Miño CI (2015) Genetic structuring among populations of the great egret, Ardea alba egretta, in major Brazilian wetlands. Aquat Conserv Mar Freshw Ecosyst 26:333–349. doi:10.1002/aqc.2588
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at H-2 gene complex. Nature 256:50–52. doi:10.1038/256050a0
Ekblom R, Saether SA, Jacobsson P, Fiske P, Sahlman T, Grahn M, Kålås JA, Höglund J (2007) Spatial pattern of MHC class II variation in the great snipe (Gallinago media). Mol Ecol 16:1439–1451. doi:10.1111/j.1365-294X.2007.03281.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Fabiani A, Hoelzel AR, Galimberti F, Muelbert MMC (2003) Long-range paternal gene flow in the southern elephant seal. Science 299:676. doi:10.1126/science.299.5607.676
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587. doi:10.3410/f.1015548.197423
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496. doi:10.1016/j.tree.2012.05.012
Germain RN, Castellino F, Han R, Sousa CRE, Romagnoli P, Sadegh-Nasseri S, Zhong G (1996) Processing and presentation of endocytically acquired protein antigens by MHC class II and class I molecules. Immunol Rev 151:5–30. doi:10.1111/j.1600-065X.1996.tb00701.x
Goudet J (1995) FSTAT, a program to estimate and test gene diversities and fixation indices (version 1.2). J Hered 86:485–486
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hughes AL (1991) MHC polymorphism and the design of captive breeding programs. Conserv Biol 5:249–251. doi:10.1111/j.1523-1739.1991.tb00130.x
Hughes AL, Nei M (1992) Maintenance of MHC polymorphism. Nature 355:402–403. doi:10.1038/355402b0
Hull JM, Anderson R, Bradbury M, Estep JA, Ernest HB (2008) Population structure and genetic diversity in Swainson’s Hawks (Buteo swainsoni): implications for conservation. Conserv Genet 9:305–316. doi:10.1007/s10592-007-9342-y
IUCN (2015) IUCN Red list of threatened species. Available via DIALOG. http://www.iucnredlist.org. Accessed 27 Jan 2015
Janeway CA, Murphy K, Travers P, Ehrenstein M (2008) Janeway’s immunobiology. Garland, New York
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:13. doi:10.1186/1471-2156-6-13
Jones MR, Cheviron ZA, Carling MD (2014) Variation in positively selected major histocompatibility complex class I loci in rufous-collared sparrows (Zonotrichia capensis). Immunogenetics 66:693–704. doi:10.1007/s00251-014-0800-7
Klareskog L, Sandgerg-Tragardh L, Rask L, Lindblom JB, Curman B, Peterson PA (1977) Chemical properties of human Ia antigens. Nature 265:248–251. doi:10.1038/265248a0
Klein J (1986) Natural history of the major histocompatibility complex. Wiley, New York
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Wakeland EK, Watkins DI (1990) Nomenclature for major histocompatibility complexes of different species: a proposal. Immunogenetics 31:217–219. doi:10.1007/978-3-642-77506-2_32
Klein J, Satta Y, O’hUigin C, Takahata N (1993) The molecular descent of the major histocompatibility complex. Annu Rev Immunol 11:269–295. doi:10.1146/annurev.iy.11.040193.001413
Kohyama TI, Omote K, Nishida C, Takenaka T, Saito K, Fujimoto S, Masuda R (2015) Spatial and temporal variation at major histocompatibility complex class IIB genes in the endangered Blakiston’s fish owl. Zool Lett 1:13. doi:10.1186/s40851-015-0013-4
Koutsogiannouli EA, Moutou KA, Stamatis C, Walter L, Mamuris Z (2014) Genetic variation in the major histocompatibility complex of the European brown hare (Lepus europaeus) across distinct phylogeographic areas. Immunogenetics 66:379–392. doi:10.1007/s00251-014-0772-7
Kushlan JA, Hancock JA (2005) The Herons. Oxford University Press, Oxford
Lanfear R, Calcott B, Ho SY, Guindon S (2012) Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701. doi:10.1093/molbev/mss020
Lei W, Fang W, Lin Q, Zhou X, Chen X (2015) Characterization of a non-classical MHC class II gene in the vulnerable Chinese egret (Egretta eulophotes). Immunogenetics 67:463–472. doi:10.1007/s00251-015-0846-1
Lei W, Zhou X, Fang W, Lin Q, Chen X (2016) Major histocompatibility complex class II DAB alleles associated with intestinal parasite load in the vulnerable Chinese egret (Egretta eulophotes). Ecol Evol 6:4421–4434. doi:10.1002/ece3.2226
Li L, Zhou X, Chen X (2011) Characterization and evolution of MHC class II B genes in ardeid birds. J Mol Evol 72:474–483. doi:10.1007/s00239-011-9446-3
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Lillie M, Grueber CE, Sutton JT, Howitt R, Bishop PJ, Gleeson D, Belov K (2015) Selection on MHC class II supertypes in the New Zealand endemic Hochstetter’s frog. BMC Evol Biol 15:63. doi:10.1186/s12862-015-0342-0
Luo M, Pan H, Liu Z, Li M (2012) Balancing selection and genetic drift at major histocompatibility complex class II genes in isolated populations of golden snub-nosed monkey (Rhinopithecus roxellana). BMC Evol Biol 12:207. doi:10.1186/1471-2148-12-207
Martínez-Cruz B, Godoy JA, Negro JJ (2004) Population genetics after fragmentation: the case of the endangered Spanish imperial eagle (Aquila adalberti). Mol Ecol 13:2243–2255. doi:10.1111/j.1365-294X.2004.02220.x
Meyers LA, Bull JJ (2002) Fighting change with change: adaptive variation in an uncertain world. Trends Ecol Evol 17:551–557. doi:10.1016/S0169-5347(02)02633-2
Miller HC, Lambert DM (2004) Genetic drift outweighs balancing selection in shaping post-bottleneck major histocompatibility complex variation in New Zealand robins (Petroicidae). Mol Ecol 13:3709–3721. doi:10.1111/j.1365-294X.2004.02368.x
Morin PA, Luikart G, Wayne RK, The SNP workshop group (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216. doi:10.1016/j.tree.2004.01.009
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152. doi:10.1146/annurev.genet.39.073003.112240
Nguyen-Phuc H, Fulton JE, Berres ME (2016) Genetic variation of major histocompatibility complex (MHC) in wild Red Junglefowl (Gallus gallus). Poult Sci 95:400–411. doi:10.3382/ps/pev364
Penn DJ, Potts WK (1999) The evolution of mating preferences and major histocompatibility genes. Am Nat 153:145–164. doi:10.1086/303166
Piertney SB (2003) Major histocompatibility complex B-LB gene variation in red grouse Lagopus lagopus scoticus. Wildl Biol 9:251–259
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21. doi:10.1038/sj.hdy.6800724
Poelstra JW, Vijay N, Bossu CM, Lantz H, Ryll B, Muller I, Baglione V, Unneberg P, Wikelski M, Grabherr MG, Wolf JBW (2014) The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 344:1410–1414. doi:10.1126/science.1253226
Promerová M, Králová T, Bryjová A, Albrecht T, Bryja J (2013) MHC class IIB exon 2 polymorphism in the Grey partridge (Perdix perdix) is shaped by selection, recombination and gene conversion. PLoS One 8:e69135. doi:10.1371/journal.pone.0069135
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. doi:10.1111/j.1471-8286.2007.01931.x
Schut E, Rivero-de Aguilar J, Merino S, Magrath MJ, Komdeur J, Westerdahl H (2011) Characterization of MHC-I in the blue tit (Cyanistes caeruleus) reveals low levels of genetic diversity and trans-population evolution across European populations. Immunogenetics 63:531–542. doi:10.1007/s00251-011-0532-x
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279. doi:10.2307/2410134
Smith S, Goüy de Bellocq J, Suchentrunk F, Schaschl H (2011) Evolutionary genetics of MHC class II beta genes in the brown hare, Lepus europaeus. Immunogenetics 63:743–751. doi:10.1007/s00251-011-0539-3
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16. doi:10.1186/1742-9994-2-16
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B 277:979–988. doi:10.1098/rspb.2009.2084
Spurgin LG, van Oosterhout C, Illera JC, Bridgett S, Gharbi K, Emerson BC, Richardson DS (2011) Gene conversion rapidly generates major histocompatibility complex diversity in recently founded bird populations. Mol Ecol 20:5213–5225. doi:10.1111/j.1365-294X.2011.05367.x
Takahata N, Nei M (1990) Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics 124:967–978
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi:10.1093/molbev/mst197
Vásquez-Carrillo C, Friesen F, Hall L, Peery MZ (2013) Variation in MHC class II B genes in marbled murrelets: implications for delineating conservation units. Anim Conserv 17:244–255. doi:10.1111/acv.12089
Wang Z, Zhou X, Lin Q, Fang W, Chen X (2013) Characterization, polymorphism and selection of major histocompatibility complex (MHC) DAB genes in vulnerable Chinese egret (Egretta eulophotes). PLoS One 8:e74185. doi:10.1371/journal.pone.0074185
Weber DS, Stewart BS, Schienman J, Lehman N (2004) Major histocompatibility complex variation at three class II loci in the northern elephant seal. Mol Ecol 13:711–718. doi:10.1111/j.1365-294X.2004.02095.x
Witzenberger KA, Hochkirch A (2011) Ex situ conservation genetics: a review of molecular studies on the genetic consequences of captive breeding programmes for endangered animal species. Biodivers Conserv 20:1843–1861. doi:10.1007/s10531-011-0074-4
Zeng Q, He K, Sun D, Ma M, Ge Y, Fang S, Wan Q (2016) Balancing selection and recombination as evolutionary forces caused population genetic variations in golden pheasant MHC class I genes. BMC Evol Biol 16:42. doi:10.10.1186/s12862-016-0609-0
Zhou X, Fang W, Chen X (2010) Mitochondrial DNA diversity of the vulnerable Chinese Egret (Egretta eulophotes) from China. J Ornithol 151:409–414. doi:10.1007/s10336-009-0470-7
Zhu Y, Wan Q, Yu B, Ge Y, Fang S (2013) Patterns of genetic differentiation at MHC class I genes and microsatellites identify conservation units in the giant panda. BMC Evol Biol 13:227. doi:10.1186/1471-2148-13-227
Acknowledgments
We thank Yufei Dai who helped collect some samples for this study. This work was funded by the National Natural Science Foundation of China (Grant Nos. 41476113 and 31272333) and by the Fujian Natural Science Foundation of China (2010Y2007).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We declare that we have no conflict of interests.
Ethical approval
All procedures involving collection of animal tissue in the wild were approved by the Administration Center for Wildlife Conservation in Fujian Province (FJWCA-1208) and were in accordance with its ethical standards.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10592_2016_876_MOESM1_ESM.doc
Fig. S1 Mean of log probability of L (K) and Delta K over 10 runs for each K value. Supplementary material 1 (DOC 1230 kb)
Rights and permissions
About this article

Cite this article
Lei, W., Fang, W., Zhou, X. et al. Population genetic diversity and geographical differentiation of MHC class II DAB genes in the vulnerable Chinese egret (Egretta eulophotes). Conserv Genet 17, 1459–1468 (2016). https://doi.org/10.1007/s10592-016-0876-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0876-8