
Abstract
Habitat fragmentation is a major threat to the maintenance of genetic diversity in many plant populations. Genetic effects of population size have received far more attention than the effects of isolation—or connectivity—but both are key components of the fragmentation process. To analyze the consequences of fragment size and connectivity on the neutral genetic variation and population genetic structure of the dominant gypsophile Lepidium subulatum, we selected 20 fragments along two continuous gradients of size and degree of isolation in a fragmented gypsum landscape of Central Spain. We used eight polymorphic microsatellite markers, and analyzed a total of 344 individuals. Populations were characterized by high levels of genetic diversity and low inbreeding coefficients, which agrees with the mainly outcrossing system of L. subulatum and its high abundance in gypsum landscapes. Bayesian clustering methods, pairwise F ST values and analysis of molecular variance revealed low among-population differentiation, with no significant isolation by distance. However, several genetic diversity indices such as allelic richness, number of effective alleles, expected heterozygosity and number of private alleles were negatively related to population isolation. The higher genetic diversity found on more connected fragments suggests higher rates of gene flow among more connected populations. Overall, our results highlight that fragmentation can have important effects on intra-population genetic processes even for locally abundant, dominant species. This, together with previously documented effects of connectivity on fitness of gypsophile species highlights the importance of including habitat connectivity in management and conservation strategies of this type of semiarid systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat fragmentation presents a major threat to biodiversity worldwide, and is one of the main driving forces behind current genetic diversity loss in many plant populations (Young et al. 1996; Lindenmayer and Fischer 2006). In arid and semi-arid Mediterranean environments, it becomes especially relevant due to the profound land use changes occurred over centuries, mainly through agriculture and afforestation practices (Pueyo et al. 2008). Habitat fragmentation reduces the size and increases the spatial isolation of populations, which can reduce genetic variation and its distribution across the landscape (Young et al. 1996; Aguilar et al. 2008). In general, habitat loss may cause a decrease in population size, which may involve risks of inbreeding depression, loss of genetic variation, fixation of deleterious alleles as well as increased population differentiation as a result of genetic drift (Wright 1951; Lande 1988; Barrett et al. 1991; Ellstrand and Elam 1993). Moreover, increased isolation (i.e., reduced connectivity) of fragmented populations can affect pollen and seed dispersal, altering patterns of gene flow and thereby genetic diversity among populations (Young et al. 1996; Sork et al. 1999; Sork and Smouse 2006).
Although both fragment size and connectivity can have important effects on the amount and distribution of genetic diversity (Aguilar et al. 2008), many studies in fragmented landscapes have focused mainly on the effects of fragment and/or population size, overlooking the effects of connectivity (see e.g. Van Rossum et al. 2004; Leimu et al. 2006; Ganzhorn et al. 2015; Raabová et al. 2015; but see Leblois et al. 2006; Honnay et al. 2007; Tamaki et al. 2008; Matesanz et al. 2015). Despite this oversimplification, it may be expected that the net effect of habitat fragmentation in a given population is a result of the interaction between different factors (Leblois et al. 2006); therefore, addressing only the effects of reductions in fragment and/or population size may not fully reflect multiple processes occurring in a fragmented landscape (Aguilar et al. 2008). For instance, moderate or even relatively low levels of gene flow among connected plant populations can mitigate the genetic effects associated with small population sizes by replenishing alleles lost through genetic drift (e.g. Schaal and Leverich 1996; Sork et al. 1999; Couvet 2002). However, experimental designs where populations are selected along continuous and independent gradients of size and degree of isolation are rather uncommon in fragmentation studies, even though they can provide robust tests of the effects of both components. Even more, partialling out the confounding effect of small population sizes should be considered to properly test the effects of fragment size. In small habitat patches, populations may have reduced genetic diversity due to low population sizes rather than to the effects of habitat size per se. This confounding factor can be minimized by selecting fragments with moderate population sizes where the deleterious effects linked to small population sizes, such as inbreeding, genetic drift and reproductive stochasticity are lower.
Plant communities established on gypsum soils constitute a compelling study system to assess the ecological, genetic and evolutionary effects of habitat fragmentation, due to their unique landscape and biological features (Escudero et al. 2015). Gypsum habitats conform naturally fragmented configurations (Martínez-Nieto et al. 2012), defined by the presence of low rounded gypsum hills of varying size or edaphic islands surrounded by flat-bottomed valleys, where different plant communities occur (Rivas-Martínez and Costa 1970). This implies that the evolutionary history of gypsum plants is intimately linked to the insular structure of the habitat (Escudero et al. 2015). This natural archipelago-like configuration has become increasingly fragmented due to agriculture intensification (Guerrero-Campo et al. 1999; Matesanz et al. 2009, 2015). Therefore, current remnants of gypsum habitats are the result of both natural and anthropic fragmentation and constitute evolutionary scenarios in which fragmentation has operated for a long time (Matesanz et al. 2009; Escudero et al. 2015). Furthermore, gypsum soils are also characterized by a diverse and specialized flora where endemic and endangered plants abound (Rzedowski 1955; Mota et al. 2004). Due to their high biodiversity and vulnerability, they have been recognized as a remarkable biodiversity hotspot in terrestrial ecosystems (Escudero et al. 2015), being designated as priority habitats for conservation by the European Union (Habitats Directive 92/43/EEC; habitat 1520). The conservation value of their flora, together with their particular landscape configuration, call for studies aimed to understand how past and ongoing fragmentation shape population genetic structure and whether it may limit future plant adaptation (Leimu et al. 2010).
Our main goal was to evaluate the consequences of fragment size and connectivity on genetic variation and population genetic structure of the dominant and abundant gypsophile Lepidium subulatum L. The study species is an Ibero-North African endemic with a wide and dominant distribution in gypsum landscapes. This small perennial shrub is a strict habitat-specialist that exclusively grows on gypsum soils (Palacio et al. 2007). The high habitat specificity of this species, together with its short-distance seed dispersal (Escudero et al. 2000, 2015), dependency on pollinators (self-incompatible; Gómez et al. 1996) and relatively short-life span (Eugenio et al. 2012), make it an ideal plant to study the genetic effects of fragmentation (Aguilar et al. 2008). Indeed, a previous study on the effects of habitat fragmentation on fitness of L. subulatum populations revealed that plants from more connected fragments had higher reproductive success via increased seed mass (Matesanz et al. 2015). Accordingly, we hypothesized that populations of L. subulatum on more isolated fragments will have lower genetic diversity and higher population differentiation than connected ones, irrespective of their size. We selected 20 fragments (populations) with different size and degree of isolation in a gypsum landscape of Central Spain, where this species grows abundantly, and addressed the following questions: (i) what is the relative importance of both fragment size and connectivity on intra-population genetic variation of L. subulatum?; (ii) are populations genetically differentiated from one another?; and (iii) is there spatial genetic structure? If so, is that structure related to fragment size and connectivity? i.e., are smaller and/or geographically more-isolated populations genetically more differentiated than larger and/or more-connected populations?
Materials and methods
Species and study site
Lepidium subulatum L. (Cruciferae) is an endemic species of the Iberian Peninsula and North Africa (Hernández-Bermejo and Clemente 1993). This perennial plant is a small (20–60 cm in height) shrub and a genuine gypsophile, i.e., a species that occurs only on gypsum soils (Palacio et al. 2007). It is a self-incompatible species (Gómez et al. 1996) with generalist entomophilous pollination that is visited by diverse pollinators, such as several Bombyliidae, Apidae, Coleoptera and Lepidoptera species (Escudero et al., unpublished data). Its seeds, very small, lack any feature to assist dispersion and they are covered by a mucilage that favors their adhesion to the gypsum surface crust (Escudero et al. 2000). Flowering peaks in May–June in Central Spain and individuals can produce ≈6000 flowers (Matesanz et al. 2015). It is a diploid species with 2n = 16 chromosomes (Martínez-Nieto et al. 2012).
This study was carried out in a gypsum landscape located 45 km southeast of Madrid, in Central Spain (40º 10′N, 3º25′W, 673 m.a.s.l.). The area has a Mediterranean semiarid climate, with an annual average rainfall of 440 mm, characterized by considerable interannual variation, a severe summer drought, and a mean annual temperature of 14 °C (Palacio et al. 2007). These dry conditions, combined with the characteristics of gypsum soils determine the presence of a very specialized vegetation, dominated by a large number of medium- to low-sized woody species. Since the 1950s, intense agricultural activities have transformed the matrix exacerbating the differences among gypsum habitats on small hills and a very adverse matrix formed by extensive crops such as vines, olive trees, cereals and Pinus plantations (Guerrero-Campo et al. 1999; Matesanz et al. 2015).
Sampling design
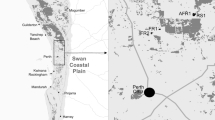
In summer 2012, we selected 20 fragments within a 5 × 6 km rectangular area, in a gypsum landscape where L. subulatum populations were present and abundant (Fig. 1). A fragment was defined as a gypsum vegetation patch, surrounded by croplands/plantations with sharp limits. Altitude, slope aspect and fragment age, i.e., factors that have been previously found to affect genetic variation (Aguilar et al. 2008), were homogenized in the selection of the fragments (see Matesanz et al. 2015 and Online Resource 1 for details on fragment selection). Lepidium subulatum populations on each fragment had a minimum of 150 individuals to tease apart the effects of fragment features from those associated with small population sizes.

Fragments of natural gypsum vegetation in the sampled area (dark grey areas). Sampled fragments are highlighted in black (N = 20) and identified by the fragment codes presented in Table 1. The selected fragments represent a size and a connectivity gradient (see text for details), and (b) distribution range of L. subulatum in the Iberian Peninsula indicated with black dots and location of the sampled area
All selected fragments created both a size and a connectivity gradient, independent from each other (Online Resource 1). Fragment area and minimum distance between fragment borders were calculated on ArcGis 10.1 (ESRI, Redlands, CA, USA), using high-resolution orthophotos of the area from 2011 (www.ign.es). Connectivity of each fragment (Ci) was calculated as an index, which takes into account the distance of the focal fragment i to all other (k) surrounding L. subulatum populations included within a 500 m radius circle (d 2 ik ), together with their fragment size (A k ) (Ci = log10 ∑ A k /d 2 ik ; see Matesanz et al. 2015 for a detailed description of the index). In the spring of 2013, we randomly selected 20 flowering individuals from each fragment in a 20 × 20 m plot, each separated by at least 1 m from its neighbors. From all selected individuals we collected young leaves and stored them in paper bags with silica gel at room temperature. In total, 400 individuals from 20 populations were collected.
DNA extraction and microsatellite markers
Genomic DNA was extracted from 60 mg of leaf tissue using the SpeedTools Plant DNA Extraction kit (Biotools, Madrid, Spain), following the manufacturer’s protocol. DNA quality and quantity were estimated using 1 % agarose gels. Eight microsatellite markers (loci), previously described for L. subulatum by Martínez-Nieto et al. (2012), were selected for their high level of polymorphism across the species: Lsub01, Lsub02, Lsub03, Lsub04, Lsub05, Lsub07, Lsub08 and Lsub12.
PCR reactions for each microsatellite marker were performed in a 20.2 µL volume containing 14.2 µL of autoclaved Milli-Q water, 2 µL of Buffer with 2 mM MgCl2 (Biotools, Madrid, Spain), 0.2 µL of each dNTP (Biotools, Madrid, Spain), 0.5 µL of the forward primer (labelled with 6FAM, NED, PET or VIC fluorescent dyes from Applied Biosystems, Madrid, Spain), 0.5 µL of the reverse (unlabelled) primer, 1 U Taq DNA polymerase (Biotools, Madrid, Spain) and 1.2 µL of template DNA. Amplification reactions were performed in a S1000 Thermal Cycler (Bio-Rad Laboratories, CA, USA) with the following cycle: initial denaturation step of 94 °C for 4 min, followed by 30 cycles of 1 min at 94 °C, 1 min with annealing temperature of 55 °C and 1 min at 72 °C, and a final extension at 72 °C for 7 min. One negative control without DNA was included in each amplification and, approximately 10 % of the samples were re-amplified, as a control, to ensure a correct genotyping. The amplified products obtained were checked by running 2.5 µL of PCR product and 2.5 µL of loading buffer (bromophenol blue, glycerol and dimethyl sulfoxide) on a 1 % agarose gel stained with GelRed (Nucleic Acid Gel Stain, 10,000 X, Biotium, USA). Successful amplifications were analyzed on an automated DNA sequencer (ABI PRISM 3730 Genetic Analyzer, Applied Biosystems, Madrid, Spain) at the Parque Científico de Madrid (http://www.fpcm.es/). Different sizes of the amplified DNA fragments were interpreted as different alleles using the GeneMarker program v. 2.61 (SoftGenetics, State College, PA, USA). Each of the eight microsatellite loci showed consistent polymorphic patterns and behaved as diploids, amplifying one or two alleles per individual. They were thus selected for genotyping of all individuals.
Data analysis
Genetic diversity within populations
For each population, we estimated the following genetic diversity indices: P 95 , the percentage of polymorphic loci by the 0.95 criterion; A, average number of alleles per locus and per population; A e , average number of effective alleles per locus and per population (1/Σp 2i , where p i is the frequency of the ith allele for the population); H o , observed heterozygosity (number of heterozygotes/N, where N is the number of individuals per population); H e , gene diversity or Nei’s unbiased expected heterozygosity ((2N/(2N − 1)) * (1 − Σp 2i )); F IS , inbreeding coefficient (1 − (H o/H e)); the number of private alleles (i.e., alleles found in a single population) and the number of multi-locus genotypes (i.e., allelic combinations of all loci examined), using the programs GenAlEx v. 6.501 (Peakall and Smouse 2012) and Genepop v. 4.2 (Rousset 2008). To obtain a conservative estimate of the number of multi-locus genotypes, we ignored the loci that could not be genotyped. Therefore, we assume that a particular multi-locus genotype is identical to another if it only differs in the loci with missing data.
Genetic diversity indices may be affected by the different number of genotyped individuals in each population. Thus, to avoid bias in the estimation of genetic diversity indices due to relatively small differences in sample size across populations, we corrected the indices that were correlated with sample size, namely allelic richness, number of private alleles and the number of multi-locus genotypes, for unequal sample sizes using the rarefaction method (Online Resource 2). Rarefaction is a statistical technique that allows to compare indexes between unequal samples (see e.g. Matesanz et al. 2014), by correcting them for the lowest sample size. As in this study the smallest sample analyzed consisted of 11 individuals (population 133, Table 1), allelic richness and the number of private alleles per locus was set to 22 (diploid species) for this calculation (A rare and A prare, respectively) and the number of multi-locus genotypes to 11 (G rare). For allelic richness and number of private alleles, this calculation was conducted using the rarefaction method implemented in the Hp-Rare software (Kalinowski 2005), and the number of multi-locus genotypes was corrected using the function rarefy implemented in the package “vegan” (Oksanen et al. 2014) for the R statistical software (v. 3.0.2) (Ihaka and Gentleman 1996).
The significance of the F IS values calculated over all loci for each population was tested with the Markov Chain Monte Carlo approximation (dememorization = 10,000, batches = 100, iterations per batch = 1000) of Fisher’s exact test implemented in Genepop v. 4.1 (Rousset 2008). In order to detect signs of recent bottlenecks or founder events, we evaluated deviations in heterozygosity from mutation-drift equilibrium in each population with the software Bottleneck 1.2.02 (Cornuet and Luikart 1996a). When a bottleneck occurs in a population and the effective population size is significantly reduced, the number of alleles decreases faster than heterozygosity, resulting in an apparent excess of heterozygosity than expected at equilibrium (Cornuet and Luikart 1996b). We performed Wilcoxon tests with 2000 iterations under three different mutation models, the stepwise-mutation model (SMM), the infinite allele model (IAM), and the intermediate two-phase mutation model (TPM) with 95 % of single-step mutations (the remaining consisting of multiple-step mutations), as recommended for microsatellites.
Finally, the effect of fragment size and connectivity on the genetic diversity indices (A rare, A e, H o, H e, F IS, A prare and G rare) was analyzed with Pearson correlations, using the software Statistica v. 8.0 (StatSoft, Tulsa, OK, USA).
Population genetic structure
We estimated the level of genetic differentiation between pairs of populations by constructing a population pairwise F ST matrix (Weir and Cockerham 1984) with P values for each pair of populations (999 permutations) using GenAlEx 6.5 software (Peakall and Smouse 2012). Significance values were corrected by the Bonferroni adjustment for a more conservative approach. The same software was used to implement a hierarchical analysis of molecular variance (AMOVA), testing its significance with 999 permutations to analyze the partitioning of molecular variance among and within populations. In order to test whether genetic differentiation among populations is related to isolation by distance, correlations between the pairwise F ST and geographic (Euclidean) distance matrices was assessed with the Mantel test based on 9999 permutations, using the GenAlEx 6.5 software (Peakall and Smouse 2012). The test was repeated using log-transformed geographical distances matrix between populations (Slatkin 1993; Rousset 1997), and similar results were obtained (see Results).
Population genetic structure was further investigated with two Bayesian clustering methods implemented in the programs Structure v. 2.3.4 (Pritchard et al. 2000; Falush et al. 2003) and Geneland v. 4.0.0 (Guillot et al. 2005). With no prior information on population sampling design, these programs probabilistically assign genotyped individuals into genetic groups or clusters where Hardy-Weinberg and linkage equilibrium within each cluster are maintained (Pearse and Crandall 2004; Guillot et al. 2005). First, we used the method implemented by the program Structure. This program assumes a model in which there are K clusters (whose number is a priori unknown), each of which is characterized by a set of allele frequencies at each locus (Pritchard et al. 2000). Individuals are then assigned to one or more clusters on the basis of their genotypes. We performed five independent runs for each K value (from 1 to 22), each run comprising a burn-in period of 105 iterations followed by 106 Markov Chain Monte Carlo (MCMC) iterations, assuming the admixture model and the model of correlated allele frequencies as recommended by Pritchard et al. (2000). To determine the most adequate numbers of clusters (K) in our data, we first analyzed the average log probability of the data for each K, and determined the value of K for which this probability was the highest. In addition, we calculated the ad hoc statistic ΔK, following the method described in Evanno et al. (2005), and identified the maximum ΔK when is plotted against K. The software Structure Harvester (Earl and vonHoldt 2012) was used to extract the relevant data from Structure result files and to generate input files for the program Clumpp. We then used Clumpp v. 1.1.2 (Jakobsson and Rosenberg 2007) to obtain the permuted membership coefficients for each individual in each cluster, combining results from the five runs at each K. We used the Greedy algorithm for K values below six and the LargeKGreedy algorithm for K values above six. The output from Clumpp was visualized in the software Distruct v. 1.1 (Rosenberg 2004).
Complementarily, we used the software Geneland, extension of the R statistical program v. 3.0.2 (Ihaka and Gentleman 1996). Geneland differs from Structure in that geographical information can be incorporated in the analysis to produce more accurate inferences of population genetic structure based on the spatial distribution of individuals (Chen et al. 2007). For the Geneland analysis, the coordinates (latitude and longitude) of each population and the genetic data of each individual were used to run the spatial and correlated allele frequency models. We performed five independent runs for each value of K ranging from 1 to 20 with the following parameters: 106 MCMC iterations, maximum rate of Poisson process fixed to 100, uncertainty attached to spatial coordinates fixed to 0.2 km, maximum number of nuclei in the Poisson-Voronoi tessellation fixed to 400 and a burn-in period of 1000. Finally, we also performed five independent runs assuming both the uncorrelated allele frequencies and non-spatial models, for comparative purposes.
Results
From the 400 individuals sampled, a total of 344 were successfully genotyped. All populations had a minimum of 14 genotyped individuals, with the exception of two populations (133 and 136) with 11 and 12 individuals, respectively (Table 1). In the 344 individuals, 8.72 % of all alleles for the eight microsatellite loci could not be determined.
Genetic diversity within populations and effects of habitat fragmentation
Genetic diversity was generally high in all populations. The eight microsatellite loci scored gave a total of 134 alleles in the 344 individuals of L. subulatum examined, with an average of 16.75 alleles per locus. All of the loci were polymorphic with 12–22 alleles per locus (Table 1). In the 344 individuals, we found a total of 341 multi-locus genotypes, i.e., practically every individual represented a unique genotype except for three individuals which had the same genotype for the eight microsatellite loci analyzed as another sampled plant (Table 1). The average number of alleles observed per locus after rarefaction ranged from 4.80 in population 136 (the least connected population) to 6.83 in population 255 (Table 1). A total of 28 private alleles, which represent 20.89 % of all alleles were found in 16 of the 20 populations. The number of private alleles per population ranged from 1 to 5 (Table 1).
Expected heterozygosity values were generally high and did not vary much across populations (from 0.646 to 0.751) (Table 1). Observed heterozygosity was higher than 0.598 in all populations and slightly lower than expected heterozygosity (Table 1). The inbreeding coefficient (F IS) was very low, ranging from -0.029 (excess of heterozygotes) in population 133 to 0.184 in population 303 (Table 1), and for some populations, it was significantly different from zero (P < 0.042). When loci were separately analyzed, each population showed highly variable F IS values across loci (Online Resource 3). Assuming the infinite allele model, seven populations deviated significantly from mutation-drift equilibrium, but no sampled population showed evidence of a recent bottleneck under any mutation model (Online Resource 4).
There was no significant effect of fragment size on the genetic diversity indices per population (Table 2). In contrast, we found a significant effect of connectivity on several genetic diversity indices: the rarefacted allelic richness and the average number of effective alleles were significantly higher in more connected fragments (Fig. 2; Table 2). Even after removing the extreme values of the correlations, they remained statistically significant (for A rare: r = 0.523; P = 0.026) or marginally significant (for A e: r = 0.4508; P = 0.060). Moreover, we found marginally significant, positive effects of connectivity on expected heterozygosity and number of private alleles (Table 2). Overall, genetic diversity of L. subulatum, as measured with the above-mentioned indices, was higher in the most connected fragments, irrespective of their fragment size (see Online Resource 5 for a visual representation of the interaction between size and connectivity). Populations on isolated fragments tended to have lower allelic richness, number of effective alleles per locus, expected heterozygosity, and number of private alleles per locus, than connected populations.

a Correlation between fragment connectivity and average number of alleles per locus with rarefaction (A rare), and b correlation between fragment connectivity and average number of effective alleles per locus (A e). Shown are mean ± SE
Population genetic structure
Population pairwise F ST values were very low yet significantly different from zero, with values always lower than 0.067 (Online Resource 6); only 15.7 % of the comparisons were moderately high, i.e., above 0.05. The AMOVA showed that 95 % of the total genetic variation in L. subulatum was found within populations. Mantel test did not reveal any significant correlation between genetic and geographical distances among pairs of populations (r = 0.055; P = 0.308).
Both Bayesian clustering methods suggested that there is no significant genetic structure in the study area, with K = 1 being the number of genetic clusters that best fits the obtained data. Accordingly, the log probability of the data did not increase with higher values of K; it remained relatively stable at first and then decreased. In the graphical representations for the smallest K values (K = 2, K = 3 and K = 4), all populations showed a high proportion of genetic admixture, since no individual was entirely assigned to any genetic cluster (Online Resource 7), confirming a weak population structure with a single genetic cluster. Using Geneland, all independent runs showed also that K = 1 was the most likely number of genetic clusters. These results were also consistent with the low levels of pairwise F ST comparisons and the results obtained in the AMOVA analyses.
Discussion
Although an effect of fragment and/or population size on genetic diversity in plant populations seems to be the norm (see e.g. Van Rossum et al. 2004; Leimu et al. 2006; Ganzhorn et al. 2015; Raabová et al. 2015 and references therein), effects of habitat connectivity on genetic diversity in fragmented landscapes have received far less attention. Our findings showed that, despite high overall levels of genetic diversity and lack of strong population differentiation, there are significant and positive effects of fragment connectivity on genetic diversity of L. subulatum populations. Furthermore, these effects are far more important than those of fragment size, when the latter is assessed independently of population size. These results concur with other studies where habitat connectivity was evaluated (see e.g. Prober and Brown 1994; Cruzan 2001; Lu et al. 2005; Honnay et al. 2007; Tamaki et al. 2008; Matesanz et al. 2015), and importantly, highlight that habitat fragmentation can have significant genetic effects on dominant, locally abundant species that are often omitted in fragmentation studies in favor of rare, endangered ones (Honnay and Jacquemyn 2007; Aguilar et al. 2008; Gaston and Fuller 2008).
Habitat connectivity can alter rates of gene flow between fragments, and subsequently modify the genetic structure and genetic diversity of plant populations (Young et al. 1996; Sork et al. 1999; Rosas et al. 2011). In our study, several genetic diversity indices such as allelic richness, number of effective alleles, and to a lesser extent, expected heterozygosity and number of private alleles were negatively related to population isolation. This likely implies that gene flow into connected L. subulatum populations is higher than gene flow into more isolated ones. In other words, we suggest that in our system, populations on connected fragments, irrespective of their size, are more likely to receive and exchange genetic material—either via pollen or seed—from surrounding populations than populations on isolated fragments. These results concur with a previous study with three coexisting gypsophiles, including L. subulatum, that reported negative effects of population isolation on several reproductive traits for the study species (Matesanz et al. 2015). Irrespective of the underlying mechanisms causing the observed patterns, lower reproductive success together with lower genetic variation in isolated populations may have consequences for the long-term population viability and evolutionary adaptation (Young et al. 1996; Aguilar et al. 2008). This can be critical in a global change context, particularly for species with restricted habitat requirements such as L. subulatum, where migration can be a very limited option (Leimu et al. 2010; Matesanz and Valladares 2014).
It is noteworthy that we found a relatively large number of private alleles (≈21 % of all alleles) mainly in the most connected and peripheral (relative to our study site) fragments, with very low allele frequencies. Generally, the presence of private alleles indicates restrictions to gene flow (Slatkin and Barton 1989). However, in a context of pollen or seed exchange among all fragments, those highly connected and located in the periphery of our study area may be more likely to receive alleles from other non-sampled populations that were included in the connectivity index, therefore becoming private in our sample.
Alongside positive connectivity effects, L. subulatum showed high intra-population genetic diversity and low inbreeding coefficients. Similar results have been reported for other gypsophiles such as Jurinea pinnata (Salmerón-Sánchez et al. 2014) and Gypsophila struthium (Martínez-Nieto et al. 2013). High observed genetic diversity and low inbreeding coefficients are consistent with the mainly outcrossing mating system of L. subulatum (Loveless and Hamrick 1984; Barrett et al. 2004). This also agrees with the results of a previous pollination experiment, showing that the number of seeds produced when pollinators were excluded was dramatically reduced compared to a control, open-pollination treatment (Gómez et al. 1996). High genetic diversity in this system can also be the result of the relative high abundance of L. subulatum in gypsum landscapes, with a large number of individuals per population (see Aguilar et al. 2008). Indeed, no population showed evidence of having been affected by recent genetic bottlenecks or founder effects. Other ecological and demographic characteristics of L. subulatum, such as a generalist pollination guild (Escudero et al., unpublished data), dense persistent seed banks (Escudero et al. 2015) and overlapping generations, can also contribute to the generally high levels of genetic diversity observed (Barrett et al. 2004; Aguilar et al. 2008).
Contrary to our expectations, the majority of the genetic variation was found within populations, as shown by the low F ST values, and the fact that Bayesian clustering methods grouped individuals from all populations in a single genetic cluster. The low among-population differentiation can be explained by the existence of gene flow (via seeds and/or pollen) even among the isolated and small fragments, although exchange rates may differ among populations. Despite the limited seed dispersal mechanisms of L. subulatum, gene flow via pollen may not be severely limited for L. subulatum. This species shows a massive flower production (≈6000 flowers per plant) that are frequently visited by diverse and abundant groups of generalist entomophilous pollinators (Escudero et al. unpublished data). Furthermore, the flowering period of L. subulatum occurs earlier than for other plants in the gypsum community, which may reduce competition for pollinators with other coexisting species.
The high prevalence in the literature of fragment and/or population size effects compared to those of connectivity could be the consequence of several factors. First, in several studies the effect of fragment and/or population size on genetic diversity could be related to spatial separation between sampled individuals within a single fragment (see e.g. Van Rossum et al. 2004; Gao 2005; Honnay et al. 2006; Ganzhorn et al. 2015; but see Galeuchet et al. 2005). It can be expected to find more intra-population genetic diversity when distance among sampled individuals in each fragment is larger, and thus the sampled area differs among sites (Wright 1943). Therefore, instead of prospecting the entire area of each fragment, we deliberately maintained an equal sampled area per fragment (a plot of 20 × 20 m) in order to avoid the genetic effects associated with the geographical separation of individuals. Second, accurate measures to quantify the degree of population isolation—or connectivity—are used rarely (Tamaki et al. 2008). In particular, when organisms lack efficient dispersal mechanisms, the degree of population isolation may be influenced by the number and size of the surrounding fragments as well as the minimum distance between them and the focal population (Tremlová and Müenzbergová 2007; Matesanz et al. 2015). So far, however, most connectivity measures often ignored the number and size of the surrounding fragments and only considered the distance to the nearest population (see e.g. Llorens et al. 2004; Galeuchet et al. 2005; Lu et al. 2005).
In conclusion, our study highlights the importance of habitat connectivity on genetic diversity, given its potential influence to alter the rates of gene flow among plant populations. More research including fragment connectivity—using realistic estimates of this parameter—are required to understand the effects of isolation on population viability and future adaptation. Our study also indicates that genetic diversity is not necessarily correlated with fragment size. While, in many cases, large sizes patches of habitat have been protected by conservation and management plans, our study reinforces the importance of surrounding populations as sinks and sources of genetic variation.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188
Barrett SCH, Kohn JR, Falk DA, Holsinger KE (1991) Genetic and evolutionary consequences of small population size in plants: implications for conservation. In: Falk DA et al (eds) Genetics and conservation of rare plants. Oxford University Press, New York, pp 3–30
Barrett SCH, Cole WW, Herrera CM (2004) Mating patterns and genetic diversity in the wild Daffodil Narcissus longispathus (Amaryllidaceae). Heredity 92:459–465
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Cornuet JM, Luikart G (1996a) Description and power analysis of genetic differentiation between populations. Genetics 163:367–374
Cornuet JM, Luikart G (1996b) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Couvet D (2002) Deleterious effects of restricted gene flow in fragmented populations. Conserv Biol 16:369–376
Cruzan MB (2001) Population size and fragmentation thresholds for the maintenance of genetic diversity in the herbaceous endemic Scutellaria montana (Lamiaceae). Evolution 55:1569–1580
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Escudero A, Iriondo JM, Olano JM, Rubio A, Somolinos RC (2000) Factors affecting establishment of a gypsophyte: the case of Lepidium subulatum (Brassicaceae). Am J Bot 87:861–871
Escudero A, Palacio S, Maestre FT, Luzuriaga AL (2015) Plant life on gypsum: a review of its multiple facets. Biol Rev 90:1–18
Eugenio M, Olano J, Ferrandis P, Martínez-Duro E, Escudero A (2012) Population structure of two dominant gypsophyte shrubs through a secondary plant succession. J Arid Environ 76:30–35
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Galeuchet DJ, Perret C, Fischer M (2005) Microsatellite variation and structure of 28 populations of the common wetland plant, Lychnis flos-cuculi L., in a fragmented landscape. Mol Ecol 14:991–1000
Ganzhorn SM, Perez-Sweeney B, Thomas WW, Gaiotto FA, Lewis JD (2015) Effects of fragmentation on density and population genetics of a threatened tree species in a biodiversity hotspot. Endanger Species Res 26:189–199
Gao L-Z (2005) Microsatellite variation within and among populations of Oryza officinalis (Poaceae), an endangered wild rice from China. Mol Ecol 14:4287–4297
Gaston KJ, Fuller RA (2008) Commonness, population depletion and conservation biology. Trends Ecol Evol 23:14–19
Gómez JM, Zamora R, Hódar JA, García D (1996) Experimental study of pollination by ants in Mediterranean high mountain and arid habitats. Oecologia 105:236–242
Guerrero-Campo J, Alberto F, Hodgson J, García-Ruiz JM, Montserrat-Martí G (1999) Plant community patterns in a gypsum area of NE Spain. I. Interactions with topographic factors and soil erosion. J Arid Environ 41:401–410
Guillot G, Mortier F, Estoup A (2005) GENELAND: a computer package for landscape genetics. Mol Ecol Notes 5:712–715
Hernández-Bermejo JE, Clemente M (1993) Lepidium. In: Castroviejo S et al (eds) Flora iberica cruciferae-monotropaceae. Real Jardín Botánico-CSIC, Madrid, pp 311–327
Honnay O, Jacquemyn H (2007) Susceptibility of common and rare plant species to the genetic consequences of habitat fragmentation. Conserv Biol 21:823–831
Honnay O, Coart E, Butaye J, Adriaens D, Van Glabeke S, Roldán-Ruiz I (2006) Low impact of present and historical landscape configuration on the genetics of fragmented Anthyllis vulneraria populations. Biol Conserv 127:411–419
Honnay O, Adriaens D, Coart E, Jacquemyn H, Roldán-Ruiz I (2007) Genetic diversity within and between remnant populations of the endangered calcareous grassland plant Globularia bisnagarica L. Conserv Genet 8:293–303
Ihaka R, Gentleman R (1996) R: a language for data analysis and graphics. J Comput Graph Stat 5:299–314
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Lande R (1988) Genetics and demography in biological conservation. Science 241:1455–1460
Leblois R, Estoup A, Streiff R (2006) Genetics of recent habitat contraction and reduction in population size: does isolation by distance matter? Mol Ecol 15:3601–3615
Leimu R, Mutikainen P, Koricheva J, Fischer M (2006) How general are positive relationships between plant population size, fitness and genetic variation? J Ecol 94:942–952
Leimu R, Vergeer P, Angeloni F, Ouborg NJ (2010) Habitat fragmentation, climate change, and inbreeding in plants. In: Ostfeld RS et al (eds) The year in ecology and conservation biology 2010. Wiley-Blackwell, New York, pp 84–98
Lindenmayer DB, Fischer J (2006) Habitat fragmentation and landscape change. An ecological and conservation synthesis. Island Press, Washington
Llorens TM, Ayre DJ, Whelan RJ (2004) Evidence for ancient genetic subdivision among recently fragmented populations of the endangered shrub Grevillea caleyi (Proteaceae). Heredity 92:519–526
Loveless MD, Hamrick JL (1984) Ecological determinants of genetic structure in plant populations. Annu Rev Ecol Syst 15:65–95
Lu YQ, Waller DM, David P (2005) Genetic variability is correlated with population size and reproduction in american wild-rice (Zizania palustris var. palustris, Poaceae) populations. Am J Bot 92:990–997
Martínez-Nieto MI, Merlo ME, Mota JF, Salmerón-Sánchez E, Segarra-Moragues JG (2012) Microsatellite loci in the gypsophyte Lepidium subulatum (Brassicaceae), and transferability to other Lepidieae. Int J Mol Sci 13:11861–11869
Martínez-Nieto MI, Segarra-Moragues JG, Merlo ME, Martínez-Hernández F, Mota JF (2013) Genetic diversity, genetic structure and phylogeography of the Iberian endemic Gypsophila struthium (Caryophyllaceae) as revealed by AFLP and plastid DNA sequences: connecting habitat fragmentation and diversification. Bot J Linn Soc 173:654–675
Matesanz S, Valladares F (2014) Ecological and evolutionary responses of Mediterranean plants to global change. Environ Exp Bot 103:53–67
Matesanz S, Escudero A, Valladares F (2009) Impact of three global change drivers on a Mediterranean shrub. Ecology 90:2609–2621
Matesanz S, Theiss KE, Holsinger KE, Sultan SE (2014) Genetic diversity and population structure in Polygonum cespitosum: insights to an ongoing plant invasion. PLoS One 9:e93217
Matesanz S, Gómez-Fernández A, Alcocer I (2015) Fragment size does not matter when you are well connected: effects of fragmentation on fitness of coexisting gypsophiles. Plant Biol 17:1047–1056
Mota JF, Sola AJ, Jiménez-Sánchez ML, Pérez-García FJ, Merlo ME (2004) Gypsicolous flora, conservation and restoration of quarries in the southeast of the Iberian Peninsula. Biodivers Conserv 13:1797–1808
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2014) vegan: Community Ecology Package. R package version 2.3-2. https://CRAN.R-project.org/package=vegan
Palacio S, Escudero A, Montserrat-Martí G, Maestro M, Milla R, Albert MJ (2007) Plants living on gypsum: beyond the specialist model. Ann Bot 99:333–343
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research. Bioinformatics 28:2537–2539
Pearse DE, Crandall KA (2004) Beyond FST: analysis of population genetic data for conservation. Conserv Genet 5:585–602
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Prober SM, Brown AHD (1994) Conservation of the grassy white box woodlands: population genetics and fragmentation of Eucalyptus albens. Conserv Biol 8:1003–1013
Pueyo Y, Alados CL, Barrantes O, Komac B, Rietkerk M (2008) Differences in gypsum plant communities associated with habitat fragmentation and livestock grazing. Ecol Appl 18:954–964
Raabová J, Van Rossum F, Jacquemart A-L, Raspé O (2015) Population size affects genetic diversity and fine-scale spatial genetic structure in the clonal distylous herb Menyanthes trifoliata. Perspect Plant Ecol 17:193–200
Rivas-Martínez S, Costa M (1970) Comunidades gipsícolas del centro de España. Anales del Instituto Botánico A.J. Cavanilles 27:193–224
Rosas F, Quesada M, Lobo JA, Sork VL (2011) Effects of habitat fragmentation on pollen flow and genetic diversity of the endangered tropical tree Swietenia humilis (Meliaceae). Biol Conserv 144:3082–3088
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rousset F (2008) GENEPOP ‘ 007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Rzedowski J (1955) Notas sobre la flora y la vegetación del estado de San Luis Potosí. II. Estudio de diferencias florísticas y ecológicas condicionadas por ciertos tipos de sustrato geológico. Ciencia (México) 15:141–158
Salmerón-Sánchez E, Martínez-Nieto MI, Martínez-Hernández F, Garrido-Becerra JA, Mendoza-Fernández AJ, Gil de Carrasco C, Ramos-Miras JJ, Lozano R, Merlo ME, Mota JF (2014) Ecology, genetic diversity and phylogeography of the Iberian endemic plant Jurinea pinnata (Lag.) DC. (Compositae) on two special edaphic substrates: dolomite and gypsum. Plant Soil 374:233–250
Schaal BA, Leverich WJ (1996) Molecular variation in isolated plant populations. Plant Species Biol 11:33–40
Slatkin M (1993) Isolation by distance in equilibrium and nonequilibrium populations. Evolution 47:264–279
Slatkin M, Barton NH (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43:1349–1368
Sork VL, Smouse PE (2006) Genetic analysis of landscape connectivity in tree populations. Landsc Ecol 21:821–836
Sork VL, Nason J, Campbell DR, Fernandez JF (1999) Landscape approaches to historical and contemporary gene flow in plants. Trends Ecol Evol 14:219–224
Tamaki I, Setsuko S, Tomaru N (2008) Genetic variation and differentiation in populations of a threatened tree, Magnolia stellata: factors influencing the level of within-population genetic variation. Heredity 100:415–423
Tremlová K, Müenzbergová Z (2007) Importance of species traits for species distribution in fragmented landscapes. Ecology 88:965–977
Van Rossum F, Campos De Sousa S, Triest L (2004) Genetic consequences of habitat fragmentation in an agricultural landscape on the common Primula veris, and comparison with its rare congener, P-vulgaris. Conserv Genet 5:231–245
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1943) Isolation by distance. Genetics 28:114–138
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418
Acknowledgments
We thank A. García-Fernández, M. Rodríguez, L. Concostrina-Zubiri, C. Díaz and G. Prieto-Porriños for their help during field and lab work, and especially A. Escudero and A. García-Fernández for their valuable comments and suggestions that contributed to improve the manuscript. Funding was provided by the British Ecological Society (research grant), the Spanish Association for terrestrial ecology (AEET, junior researcher grant), Remedinal-3 (S2013/MAE-2719) and the Juan de la Cierva Post-doctoral Program.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gómez-Fernández, A., Alcocer, I. & Matesanz, S. Does higher connectivity lead to higher genetic diversity? Effects of habitat fragmentation on genetic variation and population structure in a gypsophile. Conserv Genet 17, 631–641 (2016). https://doi.org/10.1007/s10592-016-0811-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0811-z