
Abstract
Many island avian populations are of conservation interest because they have a higher risk of extinction than mainland populations. Susceptibility of island birds to extinction is primarily related to human induced change through habitat loss, persecution, and introduction of exotic species, in combination with genetic factors. We used microsatellite profiles from 11 loci to assess genetic diversity and relatedness in the critically endangered hawk Buteo ridgwayi endemic to the island of Hispaniola in the Caribbean. Using samples collected between 2005 and 2009, our results revealed a relatively high level of heterozygosity, evidence of a recent genetic bottleneck, and the occurrence of inbreeding within the population. Pair relatedness analysis found 4 of 7 sampled breeding pairs to be related similar to that of first cousin or greater. Pedigree estimates indicated that up to 18 % of potential pairings would be between individuals with relatedness values similar to that of half-sibling. We discuss our findings in the context of conservation genetic management suggesting both carefully managed translocations and the initiation of a captive population as a safeguard of the remaining genetic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Island avian populations have a higher risk of extinction than mainland populations (Myers 1979; Smith et al. 1993) with 90 % of bird extinctions in historic times being island species (Myers 1979). Susceptibility of island birds to extinction is primarily related to human induced change through habitat loss, persecution, and introduction of exotic species (Frankham 1997; Pimm et al. 2006; Boyer 2010), in combination with genetic factors (Frankham 1998, 2005). Fluctuations in effective population sizes, genetic drift, and reduced gene flow in small, isolated populations can result in reduced genetic variation and inbreeding depression (Hedrick and Kalinowski 2000; Keller and Waller 2002; Dunn et al. 2011) leading to a greater risk of extinction as a result of lower fitness and lack of ability to adapt to rapid changes in environmental conditions (Frankham 1995a; Reed et al. 2003; Bolund et al. 2010). Most populations are impacted by genetic factors before they are driven to extinction (Spielman et al. 2004; Frankham 2005).
In general, island endemic species exhibit lower genetic variation than mainland populations (Frankham 1997). Allelic richness and heterozygosity have been lower in smaller, isolated fragmented populations than in larger more diverse populations of the same species (Johnson et al. 2004; Martinez-Cruz et al. 2004; Bollmer et al. 2005; Huang et al. 2005). Population status also appears to be associated with genetic diversity (Johnson and Stattersfield 1990). An analysis of 170 pairs of threatened and related non-threatened taxa revealed that mean heterozygosity was 35 % lower for the threatened counterparts (Spielman et al. 2004). Evans and Sheddon (2008) examined correlates of genetic diversity with respect to conservation status in 194 bird species and found a significant decline in mean heterozygosity with increasing extinction risk.
Within small isolated populations, the degree of genetic relatedness within the remaining population is an important factor to consider for conservation management planning. Inbreeding increases a species’ risk of extinction (Frankham 1995a, b), and if detected, may mean that translocations among isolated populations are needed. If genetic factors are not considered during population studies, extinction risks may be underestimated and unsuitable recovery strategies implemented (Frankham 2005). Molecular marker-based relatedness estimators can infer relationships among individuals with unknown ancestry, in the absence of pedigree information (Russello and Amato 2004), providing necessary information for developing appropriate management strategies. Implementing rational genetic management of threatened species in the wild is a top priority in conservation genetics (Frankham 2010).
Ridgway’s Hawk (Buteo ridgwayi) is a forest raptor endemic to Hispaniola in the Caribbean. The species was locally common in areas of Haiti and the Dominican Republic at the turn of the century (Cory 1885; Wetmore and Lincoln 1934), but is now listed as Critically Endangered (IUCN 2010). The current global population size is estimated at <110 pairs, limited to an area of 1,600 km2 of karst rainforest in Los Haitises National Park on the northeast coast of the Dominican Republic (IUCN 2010; Woolaver 2011). Forest loss due to slash-and-burn agriculture and human persecution of hawks have been major factors in the species decline (Woolaver 2011). Nearly all of the original forest cover has disappeared from Haiti, and 90 % of the Dominican Republic’s original forests have been destroyed by human activity (Harcourt and Ottenwalder 1996). The restricted range, small population size, and threatened status of Ridgway’s Hawk make it vulnerable to the genetics of small populations. Here we examine the population genetics and adult relatedness within the small declining Ridgway’s Hawk population to assess the extent to which genetic factors increase risk of extinction and to incorporate these findings within an appropriate conservation strategy.
Methods
Study area
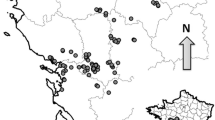
The island of Hispaniola (19°0′N; 71°0′W) is located in the Caribbean and consists of the nations of Haiti and the Dominican Republic. The cool, wet season is from April to December with eastern regions of the island receiving >2,000 mm of annual rainfall and the humid wet forests receiving the highest annual amounts at >3,000 mm. Less than 1.5 % of Haiti’s original forest is left, most of which is in the inaccessible uplands of the island and is highly degraded (Rimmer et al. 2005). The Dominican Republic has not fared much better with only 10 % of its original forest cover remaining under threat to further loss from unregulated logging, slash-and-burn agriculture, and charcoal production (Latta et al. 2006). We conducted our study in Los Haitises National Park (19ºN; 70ºW) which ranges from 0 to 380 m asl in elevation and is located on the northeast coast of the Dominican Republic (Fig. 1). It is a platform karst (eroded limestone) formation, with dense clusters of steep conical hills, or mogotes, of nearly uniform height (200–300 m) separated by sinkhole valleys. The Los Haitises region consists of thousands of such mogotes within an area of 1,600 km2.

a Maps showing relative location of Hispaniola in the Caribbean; and the island of Hispaniola with nations of Haiti and Dominican Republic and their respective capital cities: Port-au-Prince (1) and Santo Domingo (2). The study area of Los Haitises National Park boundaries are delineated in black (3). b Map of locations of all individuals sampled during the study period
Nest monitoring
Breeding pairs of Buteo ridgwayi were studied over five breeding seasons (2005–2009). Early-season observations for breeding pairs were made from vantage points on hillsides overlooking valleys to identify subsequent probable nest locations. Once found, nests were visited every 1–3 days (for easily accessible nests), or every 1–2 weeks for sites that were more difficult to access. Nestlings were banded at the nest when 25–40 days old. Nestlings were placed in cotton bags and lowered to the ground below the nest, where they were measured, banded, and a blood sample collected for DNA. Handling time of nestlings did not exceed 20 min per individual. Adults were captured using bal-chatri noose traps baited with white domestic mice Mus musculus (Thorstrom 1996). Adults were not trapped when the pair was incubating eggs.
DNA collection and extraction
Whole blood was collected from 149 Ridgway’s Hawks during the study period: 35 adults, 2 fledglings and 112 nestlings. Approximately 0.2 ml of blood was drawn via capillary tube from a patagial vein puncture, half of which was stored in 1.6 ml of Queen’s lysis buffer (Seutin et al. 1991). The other 0.1 ml of blood was stored in 1.8 ml of 95 % ethanol. All samples were stored at ambient temperature until delivered to laboratory facilities where they were preserved at −20 °C.
Total cell DNA was isolated by blood cell lysis, followed by DNA precipitation using ammonium acetate and isopropanol (L. de Sousa, B. Woolfenden and S. Tarof unpublished protocol). This involved the addition of 50 µl of blood/Queen’s lysis buffer to 600 µl of cell lysis buffer and 5 µl of ice cold Proteinase K (40 ng/µl). This solution was then incubated at 55–60 °C for 5 h and then at 37 °C overnight. Ice cold ammonium acetate (200 µl) was then added, mixed gently, and centrifuged to precipitate protein. The aqueous phase, including the dissolved genomic DNA was removed and placed in a fresh tube. Ice cold isopropanol (600 µl) was added and the solution inverted until DNA was visible as a white floating string or flake. This solution was then centrifuged to collect genomic DNA as a pellet at the bottom of the tube. The supernatant was removed and the DNA pellet washed with ice cold 70 % ethanol. This solution was then centrifuged and the ethanol then removed. This ethanol wash was repeated a second time. The tube was then left open and inverted overnight to allow the DNA pellet to dry completely. The DNA pellet was then suspended in 100–200 µl of TE buffer (10 mM Tris–HCl, 1 mM EDTA) at 37 °C for 24 h. DNA was stored at 4 °C while in use, and at −20 °C for longer-term storage.
DNA was visualised under ultraviolet radiation on a 1 % agarose test gel, pre-stained with ethidium bromide. Samples were visualised next to a MassRuler high range DNA ladder mix (Fermentas O’GeneRuler™).
Microsatellite genotyping
Fourteen microsatellite loci isolated from Common Buzzard Buteo buteo (Johnson et al. 2005) and Swainson’s Hawk Buteo swainsoni (Hull et al. 2007) were tested for examining allelic variation in B. ridgwayi. Polymerase Chain Reaction (PCR) protocols for each set of primers were optimized for B. ridgwayi using blood from 24 individuals (6 adult females, 6 adult males, and 12 nestlings) sampled in 2005–2007. Non-radioactive, fluorescently-labelled (Black, Blue and Green) microsatellite primers were provided by Integrated DNA Technologies (IDT™). Optimal reagent volumes and annealing temperatures varied by primer sets. In general, genomic DNA was amplified for each individual in 10 μl reactions containing 5.3–6.4 μl distilled water, 1.0 μl of PCR reaction buffer (10× TSG), 0.6–1.4 μl of 20 mM MgSO4, 0.4 μl of 10 mM dNTPs, 0.2 μl of fluorescently dyed 10 μM forward and reverse primers, 0.2 μl of Taq DNA polymerase (TSG), and 1 μl of DNA template (c.15 ng DNA in TE buffer). PCR reactions were carried out in an Eppendorf MasterCycler™ thermal cycler.
For the Bbu primer pairs: An initial 2 min denaturing step at 94 °C was followed by 12 cycles of 45 s at 94 °C, 45 s at the primer specific annealing temperature, and a 50 s extension step at 72 °C. This was followed by a further 22 cycles of 30 s at 89 °C, 45 s at the primer specific annealing temperature, and a 50 s extension step at 72 °C. The PCR reaction finished with a final 5 min extension step at 72 °C, and samples were then held at 4 °C until taken from the thermal cycler. Primer specific annealing temperatures were as follows: Bbu51 50 °C, Bbu 17 and Bbu 34 53 °C, Bbu46 54 °C, Bbu42 55 °C, Bbu03 56 °C, Bbu33 58 °C, and Bbu59 59 °C. For the Bsw primer pairs (Bsw107, 122, 207, 234, 310 and 324): An initial 2 min denaturing step at 94 °C was followed by 30 cycles of 30 s at 94 °C, 45 s at 58 °C, and a 45 s extension step at 72 °C. This reaction finished with a 30 min extension step at 72 °C and PCR products were then held at 15 °C until removed from the thermal cycler.
Each locus was amplified separately but since primers had been fluorescently labelled, loci were pooled post-PCR in Poolplex reactions. PCR products were visualized using a CEQ 8000™ DNA sequencer, and allele sizes were assigned using the Beckman Coulter CEQ 8000 Genetic Analysis System ™ software.
Data analysis
All genotype data was run through MICRO-CHECKER (Van Oosterhout et al. 2004, 2006) to check for typographical errors, scoring errors (large allele drop-out and stuttering), and the presence of null alleles.
Genetic diversity
The observed heterozygosity, expected heterozygosity, and number of alleles were calculated using ARLEQUIN software Version 3.1 (Excoffier et al. 2005). Tests of Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were completed in GENEPOP Version 4.1.10 (Rousset 2008). Linkage disequilibrium between all pairs of loci and tests for deviation from HWE were tested using a Markov exact test with 10,000 dememorization steps, 1,000 batches and 5,000 iterations per batch (Slatkin and Excoffier 1996; Guo and Thompson 1992). For both HWE and LD, a Bonferroni correction for multiple tests was implemented (Zar 1999).
Allelic richness was calculated in FSTAT Version 2.9.3 (Goudet 2001), which accounts for variation in sample sizes. FIS (inbreeding coefficient) was estimated in GENEPOP Version 4.1.10 (Rousset 2008) using the single locus estimates following a standard ANOVA (Weir and Cockerham 1984).
Population structure
The Bayesian program STRUCTURE Version 2.3 (Pritchard et al. 2000) was used to infer the number of discrete genetic populations (K) to assess if any individual hawks could potentially be migrants from an undiscovered subpopulation. This program identifies the K with the highest likelihood of explaining the data. We report our results in the form of the graph of the natural logarithm of the likelihood of the data (Ln P(D)) as recommended by Gilbert et al. (2012). The program was run for values of K = 1 through six using admixture and correlated allele frequency models (Falush et al. 2003), a burn-in of 100,000 iterations, and a run length of 1,000,000 iterations. The Ln P(D) statistic was averaged across 20 runs.
Signature of a recent bottleneck
We tested for an imprint of a recent genetic bottleneck using the program BOTTLENECK 1.2.02 (Cornuet and Luikart 1996; Piry et al. 1999). This method is based on the loss of rare alleles predicted in recently bottlenecked populations. Field-tests have verified its accuracy in detecting signatures of a population bottleneck (Cristescu et al. 2010). We conducted the Wilcoxon’s test for two mutational models in the program: the infinite alleles model (IAM) and the two phase model (TPM, Di Rienzo et al. 1994). According to Piry et al. (1999), the Wilcoxon’s test is the most appropriate and powerful test for fewer than 20 loci, and the TPM is the most appropriate mutational model for microsatellites. We used two different data sets for the analysis: adults only and nestlings only (one representative from each nest) to ensure no bias in the data set by combining generations.
Relatedness
Relatedness among the sampled adults of the population was carried out using the genetic software program KINGROUP v2 (Konovalov et al. 2004). Relatedness was determined by three methods: pairwise relatedness coefficients (r), pedigree relationships, and kinship reconstruction. Pairwise relatedness coefficients and pedigree relationships were used to detect relationships between closely related individuals including that of parent-offspring, full siblings and half-siblings. Kinship reconstruction was used to detect more distant genetic relationships, similar to that of distant cousins by grouping related kin. These methods were chosen for their proven accuracy in estimating relatedness among individuals and groups in a population (Gautschi et al. 2003).
Pairwise relatedness coefficients were calculated based on Queller and Goodnight (1989). This relatedness estimator ranges from −1 to +1. A positive value indicates that two individuals share more alleles by descent than expected by chance. First order relatives such as parent-offspring or full siblings will have an ‘r’ value of approximately 0.5, half-siblings an ‘r’ of 0.25, and first cousins an ‘r’ of 0.125. Unrelated individuals should have an ‘r’ value equal to or less than 0. Pairwise relatedness coefficients among all adults was compared to theoretical values using t tests.
Pedigree relationships were tested to determine the likelihood that a dyad (two individuals) shared a hypothesized pedigree relationship (the primary hypothesis), and whether the hypothesized relationship was significantly more likely than the alternative relationship (the null hypothesis). The primary hypothesis was that individuals of a dyad were parent-offspring or full siblings. The null hypothesis was that individuals of a dyad were unrelated.
In KINGROUP, Full Sibling Reconstruction (FSR) algorithms were created using a Simpson-assisted Descending Ratio (SDR) to reconstruct groups of kin by evaluating alternative partitions of the genotype dataset according to an overall likelihood (Konovalov et al. 2004). The SDR method is the most accurate for smaller number of loci, and the most robust to genotyping errors and the presence of unrelated individuals (Konovalov 2006). The primary hypothesis tested was that individuals of a dyad were at least as related as first cousins (r = 0.125) and belonged in the same subgroup. The null hypothesis was that individuals of a dyad were not related and should be split into different subgroups. This analysis tested for more distant genetic relationships.
Results
Three of the Buteo loci tested were monomorphic in all individuals, and therefore not included in analyses. Loci Bbu59 was monomorphic for 135 bp, Bbu03 for 205 bp and Bbu17 for 170 bp. The remaining 11 loci exhibited some variation (Table 1).
Genetic diversity
The analyses revealed between 4 and 19 alleles, with an average of 9.4 ± 4.1 alleles per locus (Table 2). Allele frequencies ranged from 0.0069 to 0.4960 (Supplementary material, Appendix 1). Observed heterozygosity across loci ranged from 0.521 to 0.884, with a mean observed heterozygosity of 0.732 ± 0.135 (Table 2). Expected heterozygosity across loci ranged from 0.538 (Bbu33) to 0.922 (Bsw324), with a mean expected heterozygosity of 0.780 ± 0.106 (Table 2). Seven of the loci exhibited significant deviations from HWE (Table 2). None of the loci showed any evidence of scoring errors from stuttering or large allele dropout when analyzed in Micro-Checker. For five of the loci with significant deviations from HWE (Bbu34, Bsw107, Bsw122, Bsw234 and Bsw310) there was no evidence of null alleles. There was evidence of possible null alleles in Bsw207 and Bsw324, and therefore allele frequencies and genotypes were adjusted in Micro-Checker to account for this. After adjustment, there were still significant deviations from HWE in the locus Bsw324 (P < 0.00), but not in Bsw207 (P = 0.59, Table 2). After Bonferroni correction, there was evidence of linkage disequilibrium for 29 of the 55 pairwise combinations of loci (Table 3). Nine of the 11 loci exhibited FIS values above zero (Table 2).
Population structure
STRUCTURE analyses indicated the best K = 1 (Fig. 2) indicating that there was no structure evident in the population and that the samples comprised a single genetic population.

Plot of mean (±SD) penalized log likelihoods of STRUCTURE analyses for K = 1 through six
Signature of a recent bottleneck
Under both the IAM and TPM, the Wilcoxon sign-rank test showed a significant excess of heterozygotes for adults (P = 0.01 IAM; P = 0.03 TPM) indicating a recent genetic bottleneck. In addition the allele frequency distribution test was a Shifted distribution further suggesting a recent bottleneck. For nestlings, there was also significant excess of heterozygotes for the IAM and TPM models (P = 0.01 IAM; P = 0.01 TPM). However, the distribution of allele frequencies was L-shaped for nestlings. When combined, the allele frequency distribution for adults and nestlings fell somewhere between the Shifted and L–shaped distribution (Fig. 3). Overall, the genetic data indicated that the Ridgway’s population has experienced a recent genetic bottleneck.

Allele frequency distribution for all loci for sampled adults and nestlings (one representative per nest)
Relatedness of adults
Relatedness coefficients of the seven sampled breeding pairs ranged from −0.09 to 0.55 and averaged 0.14 ± 0.22 (Table 4). Four of the sampled breeding pairs exhibited pairwise relatedness values that indicated a relatedness equal to or greater than first cousins. One pair was closely related similar to a full-sibling or parent-offspring relationship (r = 0.56). A second pair exhibited a relatedness coefficient of 0.21 indicating a potential half-sibling relationship. The remaining two pairs had relatedness coefficients of 0.12 and 0.15, equivalent to that of first cousins.
Relatedness coefficients of potential breeding pairs (Female–Male dyads) ranged from −0.45 to 0.62 and averaged 0.03 ± 0.18 (n = 273). This average relatedness was not significantly different from zero (t 1, 273 = −1.45, P = 0.17) indicating that, on average, matings among the sampled adults would not be between closely related individuals. However, over half the relatedness coefficients of all female–male dyads were greater than 0, and a pedigree analysis indicated that 18 % of random dyads would have relatedness values similar to that of half-siblings or greater (r > 0.20) (Fig. 4). The seven breeding pairs sampled during the study period may potentially be more related to each other, on average, than randomly paired individuals, as suggested by Fig. 4.

Pairwise relatedness coefficients (r) for all female–male dyads. The seven sampled breeding pairs are denoted as black triangles
To determine if any patterns of genetic relatedness existed within either gender, relatedness coefficients were also compared among female dyads and among male dyads. Relatedness among the 20 adult females ranged from −0.46 to 0.45 and averaged 0.09 ± 0.17. The average relatedness value was significantly different from zero (t 1,189 = 1.06, P = 0.03) indicating that the adult females sampled in this study were, on average, somewhat more related to each other than expected by chance. Relatedness among the 14 adult males sampled ranged from −0.59 to 0.52 and averaged −0.08 ± 0.16. The average relatedness value was significantly different from zero (t 1,90 = −4.16, P < 0.00). However the significance was negative indicating that, on average, adult males sampled in this study were more unrelated to each other than expected by chance (negative relatedness coefficients). Relatedness within the adult female dyads was found to be significantly higher than relatedness within the adult male dyads (t 1,279 = −2.54, P = 0.01).
Kinship reconstruction found that the 34 sampled adults consisted of 10 kin subgroups, with only three adults placed in lone groups and not related to the others (Table 4). All other adults were grouped with at least one other adult that was as related as a first cousin. The largest subgrouping contained six related adults. There did not appear to be any obvious pattern toward either sex or area in any of the kin subgroups (Table 4).
An analysis of spatial genetic orientation found no correlation between pairwise relatedness values and distance between sampled individuals for all female–male dyads (r 2 = 0.001, P = 0.599; Fig. 5a). There was also no correlation between distance and relatedness for female dyads (r 2 = 0.007, P = 0.249; Fig. 5b) or male dyads (r 2 = 0.004, P = 0.538; Fig. 5c).

Pairwise relatedness coefficients (r) in relation to distance between sampled individuals for a female–male dyads b female dyads and c male dyads
Offspring survival and homozygosity in sampled breeding pairs
Of the seven breeding pairs sampled, eight nests from six pairs were followed through to fledging of the young. The four pairs with pairwise relatedness values >0.12 produced an average of 1.0 ± 1.0 fledglings per nest (n = 5 nests), with a hatching success of 63 %. By comparison, the two unrelated pairs averaged 2.0 ± 1.0 fledglings per year (n = 3 nests), with 100 % hatching success.
The nestlings from the four related pairings also appeared to exhibit more homozygous genotypes than nestlings from two pairings that were not related. On average, the eight nestlings from inbred pairings were homozygous for 4.3 ± 1.8 (range: 3–7) of the 11 loci. By comparison, six nestlings produced by the two unrelated pairings were homozygous for an average of 1.5 ± 1.0 (range: 0–3) of the 11 loci.
Discussion
Genetic diversity
In general, Ridgway’s Hawk appear to have relatively high levels of heterozygosity (H e = 0.73) compared to other raptor species reported in the literature, including both common raptor species (Northern Goshawk Accipiter gentilis H e = 0.71 in Topinka and May 2004, Common Kestrel Falco tinnunculu H e = 0.40 in Nesje et al. 2000, Merlin Falco columbarius H e = 0.58 in Nesje et al. 2000) and threatened raptor species (Lesser Kestrel Falco naumanni at 0.70 in Hille et al. 2003, Spanish Imperial Eagle Aquila adalberti at 0.52 in Martinez-Cruz et al. 2002, Peregrine Falcon Falco peregrinus H e = 0.54 in Brown et al. 2007). The most extreme cases of island raptor declines, the contemporary populations of Mauritius Kestrel Falco punctatus and Seychelles Kestrel Falco araea exhibit extremely low levels of heterozygosity: 0.100 and 0.104 respectively (Groombridge et al. 2000, 2009). Both these populations declined to a handful of individuals prior to population recovery (Groombridge et al. 2001, 2009).
Within the genus Buteo, heterozygosity for Ridgway’s Hawk appears to be greater than Common Buzzard B. buteo (H e = 0.53 in Johnson et al. 2005) and the island populations of Galapagos Hawks (H e = 0.50 in Bollmer et al. 2011), and less than Swainson’s Hawk B. swainsoni (H e = 0.82 in Hull et al. 2008). A comparison of the homologous loci from the two species from which the primers were developed revealed that B. ridgwayi exhibits reduced levels of genetic diversity compared to both mainland species. Two homologous loci for which variability was tested on B. buteo (Bbu42 and Bbu46) exhibited higher levels of heterozygosity at 0.93 and 0.89, and higher levels of allelic diversity at 15 and 17 alleles per locus, compared to B. ridgwayi with heterozygosity of 0.79 and 0.82, and allelic diversity of 8 and 9 alleles per locus, respectively. Similarly, six homologous loci from B. swainsoni showed both higher heterozygosity levels (0.91 ± 0.04) and higher allelic diversity levels (18.5 ± 8.7 alleles per locus) compared to B. ridgwayi (heterozygosity of 0.84 ± 0.05; and allelic diversity of 11.4 ± 4.0). These results show that B. ridgwayi has reduced allelic diversity and lower heterozygosity compared to mainland Buteo species.
The appearance of relatively higher levels of heterozygosity for B. ridgwayi relative to other raptor species may be due to the fact that high levels of heterozygosity are expected for a species that has had a relatively large population size until recently, with heterozygosity decreasing slowly after a bottleneck depending on the N e (Wright 1931). In addition, since allelic diversity is reduced faster than heterozygosity during bottlenecks, this can result in transient elevated levels of heterozygosity (Nei et al. 1975).
Signature of a recent bottleneck
The results provide compelling evidence that Ridgway’s Hawk population have recently gone through a genetic bottleneck, particularly since it was apparent in both adults and nestlings. There were also many low frequency alleles observed of intermediate size, represented by only a few copies. A continued decline in the Ridgway’s Hawk population could cause the loss of these intermediate-size alleles.
A significant amount of LD was found among loci pairwise combinations. LD can be caused by several factors including population structure, hybridization, or genetic drift in small populations (Epperson and Allard 1987; Gaut and Long 2003). We found no evidence of population structure in the present study, and there is little potential for hybridization. Thus the finding of LD provides further support of a recent bottleneck in the population.
Population bottlenecks can purge low frequency alleles from a population at a rapid rate. Bellinger et al. (2003) found a 29 % loss (16/55) of alleles across six loci during a 50 year period in the Greater Prairie Chicken Tympanuchus cupido. Addison and Diamond (2011) found loss of alleles in 2 of 4 microsatellites over a 15 year period in the endemic insular Nihoa Millerbird Acrocephalus familiaris kingi. Our results indicate that the remaining population of Ridgway’s Hawks has already lost alleles during a genetic bottleneck, at an unknown rate. Thus, if the Ridgway’s population continues to decline we would expect further losses of low frequency alleles, with some alleles dropping out entirely.
Inbreeding and relatedness
The relatedness analysis verified that inbreeding does occur within the population of Ridgway’s Hawk. It also suggests that a relatively high percentage (18 %) of random pairings would be between individuals with relatedness similar to that of half-siblings. Close inbreeding has been recorded in wild populations of American Kestrel (Falco sparverius, Stewart et al. 2007), Merlin (F. columbarius, James et al. 1987), Peregrine Falcon (F. peregrinus, Tordoff and Redig 1999), Osprey (Pandion haliaetus, Postupalsky 1989) and Cooper’s Hawk (Accipiter cooperii, Rosenfield and Bielefeldt 1992; Stewart et al. 2007). These reports represent rare occurrences within large mainland populations. Assuming our sampling efforts are representative of the population, inbreeding is not a rare occurrence within the remaining Ridgway’s Hawk population. In laboratory situations, controlled experiments have shown a significantly higher extinction risk of inbred lines when compared to outbred lines (Frankham 1995a, b; Bijlsma et al. 2000; Reed et al. 2002, 2003; Wright et al. 2008). These studies indicate an increased extinction risk with not only full-sib inbreeding but also with lower orders of inbreeding.
Both immigration and dispersal have been shown to reduce the occurrence of inbreeding in bird populations (Lebigre et al. 2010; Ludwig and Becker 2012). Although natal dispersal dynamics for Ridgway’s Hawk are not known, females are generally reported to disperse further than males in raptor species (Newton 1979). Our findings that females within the study area were more related to each other than by chance may suggest either that female Ridgway’s Hawks are philopatric, or that they currently have limited dispersal opportunities. Los Haitises National Park (1,600 km2) is the last refuge for Ridgway’s Hawk, and there is very limited surrounding suitable habitat for juvenile dispersal (Woolaver 2011). If dispersal and immigration were historically the main active means to avoid inbreeding for the species without any type of kin recognition mechanisms (as has been found for other bird populations, Szulkin and Sheldon 2008; Lebigre et al. 2010; Ludwig and Becker 2012) then there may be currently few or no means of preventing inbreeding within the small remaining population. This may further explain why there was no correlation found between distance and relatedness during the study.
The concern with inbreeding is the production of offspring with increased homozygosity. This reduced genotypic variation can lead to a decrease in fitness, a process referred to as inbreeding depression (Frankham 2005). Inbreeding depression can significantly reduce the survival of offspring (Hemmings et al. 2012) and have a negative effect on reproductive success (Billing et al. 2012). Blomqvist et al. (2010) found strong evidence of inbreeding depression in the form of reduced hatching success and increased embryonic mortality, in a population of endangered shorebird Calidris alpina schinzii with a 9–13 % inbreeding rate. The remaining population of Ridgway’s Hawk appears to have a similar or potentially higher inbreeding rate, however, it was not possible to confirm with our data whether inbreeding was contributing to reduced offspring survival or reduce reproductive rates. The few data available from the sampled pairings did suggest a potential decreased in fledging success and increased homozygosity in the offspring of inbred pairs. Given that increased levels of homozygosity have been associated with increased inbreeding depression and reduced survival (Blomqvist et al. 2010), this potential toward homozygosity of nestlings needs to be investigated further and considered during management planning.
We could find no other similar studies on inbreeding or relatedness for the three other endangered Buteo species, or other wild hawk populations. Within a reintroduced population of Mauritius Kestrel (n = 199 pairs) 25 % of breeding pairs were found to consist of closely or moderately related individuals. The authors noted that conservation intervention would be required at this rate of inbreeding to prevent the fixation of detrimental alleles (Ewing et al. 2010).
Conservation implications
By combining different population genetic analysis we were able to provide a relatively broad picture of the genetic status of the remaining population of Ridgway’s Hawk. Genetic diversity (heterozygosity) appeared to be relatively high for an insular threatened species with a low population size. This could be positive for the recovery of the species, if the current population decline is halted and the population is able to increase rapidly. However, other findings from the population genetic analysis were cause for concern. In addition to habitat loss and human persecution of Ridgway’s Hawk (see Woolaver 2011), genetic factors appear to be a further threat to the species as a result of a recent genetic bottleneck and levels of inbreeding occurring within the remaining population. Conservation planning for Ridgway’s Hawk should be multifaceted and focus on several concurrent strategies prioritizing community education and awareness, and habitat protection within Los Haitises National Park. To ensure the remaining population genetic diversity is not lost further, we recommend two genetic management strategies to prevent further inbreeding and maintain current diversity levels:
-
1.
Establishment of a captive population. There appears to be no need yet for a large-scale captive breeding and re-introduction program for Ridgway’s Hawk, particularly if it were to take away from attempting to save the species within its last remaining habitat of Los Haitises. However, since the entire world population is located within one small area, the establishment of a safe guard population is highly recommended since a single small population is much more vulnerable to stochastic events and human disturbance. A properly managed captive population could retain current genetic diversity before inbreeding increases in the wild population. Selection of founder nestlings for the captive population should be carried out to identify the most genetically divergent individuals, and if possible, focus primarily on breeding pairs in areas that would otherwise face a high probability of nest failure (see Woolaver 2011).
-
2.
Translocation of selected juveniles. Conservation organisations have begun translocating juveniles from Los Haitises to establish peripheral safety net populations outside of the National Park (Peregrine Fund unpubl data). To best address the results of our population genetics analysis, future translocations should ensure that nestlings selected for translocation are genetically diverse, and if possible, chosen from areas where nests would otherwise face a high probability of failure (see Woolaver 2011). Relatedness between individuals should be an important factor taken into consideration, to ensure that genetically related juveniles (in particular genetically related birds of the opposite sex) are not translocated into the same area. In addition to the opportunity translocations allow for genetic management, releases of nestlings to other fragmented forest in the species’ historical range could also provide a catalyst for local awareness, and habitat restoration and protection outside of Los Haitises.
References
Addison JA, Diamond AW (2011) Population genetics and effective population size of the critically endangered Nihoa Millerbird (Acrocephalus familiaris kingi). Auk 128(2):265–272
Bellinger MR, Johnson J, Toepfer J, Dunn P (2003) Loss of genetic variation in greater prairie chickens following a population bottleneck in wisconsin, USA. Conserv Biol 17(3):717–724
Bijlsma R, Bundgaard J, Boerema AC (2000) Does inbreeding affect the extinction risk of small populations? predictions from Drosophila. J Evol Biol 13:502–514
Billing AM, Lee AL, Skjelseth S, Borg AA, Hale MC, Slate J, Parn H, Ringsby TH, Sæther BE, Jensen H (2012) Evidence of inbreeding depression but not inbreeding avoidance in a natural house sparrow population. Mol Ecol 21:1487–1499
Blomqvist D, Pauliny A, Larsson M, Flodin LA (2010) Trapped in the extinction vortex? strong genetic effects in a declining vertebrate population. BMC Evol Biol 10:33
Bollmer JL, Whiteman NK, Cannon MD, Bednarz JC, de Vries T, Parker PG (2005) Population genetics of the Galápagos hawk (Buteo galapagoensis): genetic monomorphism within isolated populations. Auk 122:1210–1224
Bollmer JL, Hull JM, Ernest HB, Sarasola JH, Parker PG (2011) Reduced MHC and neutral variation in the Galapagos hawk, an island endemic. BMC Evol Biol 11:143
Bolund E, Martin K, Kempenaers B, Forstmeier W (2010) Inbreeding depression of sexually selected traits and attractiveness in the zebra finch. Anim Behav 79:947–955
Boyer AG (2010) Consistent ecological selectivity through time in Pacific Island avian extinctions. Conserv Biol 24(2):511–519
Brown JW, Van Coeverden de Groot PJ, Birt T, Seutin G, Boag P, Friesen V (2007) Appraisal of the consequences of the DDT-induced bottleneck on the level and geographic distribution of neutral genetic variation in Canadian peregrine falcons, Falco peregrinus. Mol Ecol 16:327–343
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cory CB (1885) The birds of Haiti and San Domingo. Estes and Lauriat, Boston
Cristescu R, Sherwin WB, Handasyde K, Cahill V, Cooper DW (2010) Detecting bottlenecks using BOTTLENECK 1.2.02 in wild populations: the importance of the microsatellite structure. Conserv Genet 11:1043–1049
Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Dunn SJ, Clancey E, Waits LP, Byers JA (2011) Inbreeding depression in pronghorn (Antilocapra americana) fawns. Mol Ecol 20:4889–4898
Epperson BK, Allard RW (1987) Linkage disequilibrium between allozymes in natural populations of lodgepole pine. Genetics 115:341–352
Evans SR, Sheddon BC (2008) Interspecific patterns of genetic diversity in birds: correlations with extinction risk. Conserv Biol 22(4):1016–1025
Ewing SR, Nager RG, Nicoll MA, Aumjaud A, Jones CG, Keller LF (2010) Inbreeding and loss of genetic variation in a reintroduced population of Mauritius Kestrel. Conserv Biol 22(2):395–404
Excoffier LG, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587
Frankham R (1995a) Conservation genetics. Annu Rev Genet 29:305–327
Frankham R (1995b) Inbreeding and extinction: a threshold effect. Conserv Biol 9(4):305–327
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Frankham R (1998) Inbreeding and extinction: island populations. Conserv Biol 12:665–675
Frankham R (2005) Genetics and extinction. Biol Conserv 12:131–140
Frankham R (2010) Where are we in conservation genetics and where do we need to go? Conserv Genet 11:661–663
Gaut BS, Long AD (2003) The low down on linkage disequilibrium. Plant Cell 15:1502
Gautschi B, Gwenaël J, Negro JJ, Godoy JA, Müller JP, Schmid B (2003) Analysis of relatedness and determination of the source of founders in the captive bearded vulture Gypaetus barbatus population. Conserv Genet 4:479–490
Gilbert KJ, Andrew RL, Bock DG, Franklin MT, Kane NC, Moore JB, Moyers BT, Renaut SB, Rennison DJ, Veen T, Vines TH (2012) Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program STRUCTURE. Mol Ecol 21:4925–4930
Goudet J (2001) FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www.unil.ch/izea/softwares/fstat.html. Accessed 12 July 2010
Groombridge JJ, Jones CG, Bruford MW, Nichols RA (2000) ‘Ghost’ alleles of the Mauritius kestrel. Nature 403:616
Groombridge JJ, Bruford MW, Jones CG, Nichols RA (2001) Estimating the severity of the population bottleneck in the Mauritius kestrel Falco punctatus from ringing records using MCMC estimation. J Anim Ecol 70:401–409
Groombridge JJ, Dawson DA, Burke T, Prys-Jones R, de L Brooke M, Shah N (2009) Evaluating the demographic history of the Seychelles kestrel (Falco area): genetic evidence for recovery from a population bottleneck following minimal conservation management. Biol Conserv 142:2250–2257
Guo SW, Thompson EA (1992) Performing the exact test of Hardy–Weinberg proportion for multiple alleles. Biometrics 48:361–372
Harcourt C, Ottenwalder JA (1996) Hispaniola. In: Harcourt CS, Sayer JA (eds) The conservation atlas of tropical forests, the Americas. IUCN, Gland, pp 102–111
Hedrick PW, Kalinowski ST (2000) Inbreeding depression in conservation biology. Annual Rev Ecol Syst 31:139–162
Hemmings NL, Slate J, Birkhead TR (2012) Inbreeding causes early death in a passerine bird. Nat Commun 3:863
Hille SM, Nesje M, Segelbacher G (2003) Genetic structure of kestrel populations and colonization of the Cape Verde archipelago. Mol Ecol 12:2145–2151
Huang Z, Liu N, Zhou T (2005) A comparative study of genetic diversity of peripheral and central populations of Chukar partridge from northwestern China. Biochem Genet 43:613–621
Hull JM, Tufts D, Topinka JR, May BP, Ernest HB (2007) Development of 19 microsatellite loci for Swainson’s Hawks (Buteo swainsoni) and other Buteos. Mol Ecol Notes 7:346–349
Hull JM, Anderson JR, Bradbury M, Estep JA, Ernest HB (2008) Population structure and genetic diversity in Swainson’s Hawks (Buteo swainsoni): implications for conservation. Conserv Genet 9:305–316
IUCN [online] (2010) IUCN Red List of Threatened Species. Version 2010.2. www.iucnredlist.org. Accessed 12 Nov 2012
James PC, Oliphant LW, Warkentin IG (1987) Close inbreeding in the Merlin (Falco columbarius). Wilson Bull 99:718–719
Johnson TH, Stattersfield AJ (1990) A global review of island endemic birds. Ibis 132:167–180
Johnson JA, Bellinger MR, Toepfer JE, Dunn P (2004) Temporal changes in allele frequencies and low effective population size in greater prairie-chickens. Mol Ecol 13:2617–2630
Johnson PCD, Fowlie MK, Amos W (2005) Isolation of microsatellite loci from the common buzzard, Buteo buteo (Aves : accipitridae). Mol Ecol Notes 5:208–211
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Konovalov D (2006) Accuracy of four heuristics for the full sibship reconstruction problem in the presence of genotype errors.In: Jiang T, Yang U, Chen P and Wong L (eds) Proceedings of 4th Asia-Pacific bioinformatics conference. Imperial College Press, London, pp 7–16
Konovalov DA, Manning C, Henshaw MT (2004) KINGROUP: a program for pedigree relationship reconstruction and kingroup assignments using genetic markers. Mol Ecol Notes 4:779–782
Latta SC, Rimmer C, Keith A, Wiley J, Raffaele H, Mcfarland K, Fernandez E (2006) Birds of the Dominican Republic and Haiti. Princeton University Press, Princeton
Lebigre C, Alatalo RV, Siitari H (2010) Female-biased dispersal alone can reduce the occurrence of inbreeding in black grouse (Tetrao tetrix). Mol Ecol 19:1929–1939
Ludwig SC, Becker PH (2012) Immigration prevents inbreeding in a growing colony of a long-lived and philopatric seabird. Ibis 154:74–84
Martinez-Cruz B, David VA, Godoy JA, Negro JJ, O’Brien SJ, Johnson WE (2002) Eighteen polymorphic microsatellite markers for the highly endangered Spanish imperial eagle (Aquila adalberti) and related species. Mol Ecol Notes 2:323–326
Martinez-Cruz B, Godoy JA, Negro JJ (2004) Population genetics after fragmentation: the case of the endangered Spanish imperial eagle (Aquila adalberti). Mol Ecol 13:2243–2255
Myers N (1979) Islands of conservation. New Scientist 83:600–602
Nei M, Maruyama T, Chakraborty R (1975) Bottleneck effect and genetic variability in populations. Evolution 29(1):1–10
Nesje M, Roed KH, Lifjeld JT, Lindberg P, Steen OF (2000) Genetic relationships in the peregrine falcon (Falco peregrinus) analysed by microsatellite DNA markers. Mol Ecol 9:53–60
Newton I (1979) Population ecology of raptors. Buteo Books, Vermillion
Pimm SL, Raven P, Peterson A, Sekercioglu CH, Erlich PR (2006) Human impacts on the rates of recent, present, and future bird extinctions. Proc Natl Acad Sci USA 103:10941–10946
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a program for detecting recent effective population size reductions from allele data frequencies. J Heredity 90:502–503
Postupalsky S (1989) Inbreeding in Ospreys. Wilson Bull 101:124–126
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 43:258–275
Reed DH, Briscoe DA, Frankham R (2002) Inbreeding and extinction: the effect of environmental stress and lineage. Conserv Genet 3:301–307
Reed DH, Lowe EH, Briscoe DA, Frankham R (2003) Inbreeding and extinction: effects of rate of inbreeding. Conserv Genet 4:405–410
Rimmer CC, Townsend JM, Townsend AK, Fernandez EM, Almonte J (2005) Avian diversity, abundance, and conservation status in the Macaya Biosphere Reserve of Haiti. Ornitol Neotropica 16:219–230
Rosenfield RN, Bielefeldt J (1992) Natal dispersal and inbreeding in the Cooper’s Hawk. Wilson Bull 104:182–184
Rousset F (2008) Genepop’007: a complete reimplementation of the GENEPOP software for Windows and Linux. Mol Ecol Resources. 8:103–106
Russello MA, Amato G (2004) Ex-situ population management in the absence of pedigree information. Mol Ecol 13:2829–2840
Seutin G, White BN, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69:82–90
Slatkin M, Excoffier L (1996) Testing for linkage disequilibrium in genotypic data using the expectation-maximization algorithm. Heredity 76:377–383
Smith FDM, May RM, Pellew R, Johnson TH, Walter KR (1993) How much do we know about the current extinction rate? Trends Ecol Evol 8:375–378
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci USA 101:15261–15264
Stewart AC, Rosenfield RN, Nyhof NA (2007) Close inbreeding and related observations in Cooper’s hawks. J Raptor Res 41(3):227–230
Szulkin M, Sheldon BC (2008) Correlates of the occurrence of inbreeding in a wild bird population. Behav Ecol. doi:10.1093/beheco/arn086
Thorstrom R (1996) Methods for trapping tropical forest birds of prey. Wildlife Soc Bull 24(3):516–520
Topinka JR, May B (2004) Development of polymorphic microsatellite loci in the northern goshawk (Accipiter gentilis) and cross-amplification in other raptor species. Conserv Genet 5:861–864
Tordoff HB, Redig PT (1999) Close inbreeding in Peregrine Falcons in midwestern United States. J Raptor Res 33:326–327
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Van Oosterhout C, Weetman D, Hutchinson WF (2006) Estimation and adjustment of microsatellite null alleles in non-equilibrium populations. Mol Ecol Notes 6:255–256
Weir BS, Cockerham CC (1984) Estimating F-statistics for analysis of population structure. Evolution 38:1358–1370
Wetmore A, Lincoln FC (1934) Additional notes on the birds of Haiti and the Dominican Republic. Proc US Nat Mus 82:1–68
Woolaver LG (2011) Ecology and conservation genetics of Ridgway’s Hawk Buteo ridgwayi. PhD Dissertation. York University, Toronto, Canada
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright LI, Tregenza T, Hosken DJ (2008) Inbreeding, inbreeding depression and extinction. Conserv Genet 9:833–843
Zar JH (1999) Biostatistical analysis, 4th edn. Prentice Hall, Upper Saddle River
Acknowledgments
We would like to thank the following people for their advice during the study: Bonnie Woolfenden, Scott Tarof, Nigel Collar, James Wiley, Russell Thorstrom, Steve Latta, David Wege, Joel Shore, and Laurence Packer. A number of people provided significant contributions to this study particularly with searching for hawks including Jesús Almonte, Jennie Sinclair, John Vetter, Juan Cespedes, Nicolas Corona, Samuel Balbuena de la Rosa, Timoteo Bueno Hernandez, Pastor de Leon Franco and Hilario Jorge Polanco. Eladio Fernández, Jorge Brocca, and the Sociedad Ornitólogica Hispaniola provided much appreciated logistical support. All research was carried out with the permission of Subsecretaría de Estado de Areas Protegidas y Biodiversidad, and the Secretaría de Estado de Medio Ambiente y Recursos Naturales, Republica Dominicana. The research was funded by Wildlife Preservation Canada, the Natural Sciences and Engineering Research Council of Canada (NSERC) and the James Bond Fund of the Smithsonian Institution. We would particularly like to thank Elaine Williams and Kate Wallace for their support throughout the study, and Amro Zayed for guidance during analysis and for improving this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Woolaver, L.G., Nichols, R.K., Morton, E.S. et al. Population genetics and relatedness in a critically endangered island raptor, Ridgway’s Hawk Buteo ridgwayi . Conserv Genet 14, 559–571 (2013). https://doi.org/10.1007/s10592-013-0444-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-013-0444-4