
Abstract
The otton frog (Babina subaspera) is an endangered species endemic to the Amami Islands, Japan. High predation pressure from an introduced carnivore, the mongoose, has caused declines in the frog populations and created a large habitat gap around an urban area. To promote effective conservation, we investigated the genetic status of the species and examined the effect of the habitat gap on gene flow among populations. Using five polymorphic microsatellite loci and mitochondrial DNA sequences, we investigated genetic diversity, genetic structure and gene flow in B. subaspera populations on the islands of Amami-Oshima and Kakeroma-jima. The expected heterozygosity (H E) within each locality was generally high (range: 0.67–0.85), indicating that B. subaspera maintains high genetic diversity. However, genetic differentiation was observed, and the two populations, TAG and KAR, showed little gene flow with other populations. The clustering and F ST analyses also predicted that these two populations were clearly distinct. According to the mitochondrial DNA analysis, the observed genetic differentiation occurred relatively recently. Possible barriers such as mountain ridges, rivers or roads did not result in genetic separation of the populations. These data support the hypothesis that the habitat gap created by an introduced predator prevented the gene flow among B. subaspera populations. When developing conservation strategies for B. subaspera, focus should be directed to these two isolated populations; careful monitoring of population size and genetic diversity should be conducted along with the mongoose elimination project ensues.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Island populations, particularly those of endemic species, are often at high risk of extinction and require extra care for their conservation (Smith et al. 1993; Vitousek 1988). Island populations tend to be small and isolated and may have gone through population bottlenecks at the time of their founding (Frankham 1998). The small population sizes and/or population bottlenecks lead to inbreeding, which has critically negative effects on the maintenance of genetic variability (Frankham 1997; 1998; Newman and Pilson 1997). Lower genetic variation is predicted to result in inbreeding depression, reducing the fitness as well as evolutionary potential of the population (Frankham 1995). Habitat fragmentation is one factor that increases the extinction risk to island species. Habitat fragmentation often occurs when landscapes or human activities create dispersal barriers and reduce gene flow among subpopulations, which increases the risks of a decline in genetic variation caused by genetic drift and inbreeding (Andersen et al. 2004). Organisms with limited dispersal ability are especially vulnerable to these deleterious effects (Funk et al. 2005) as gene flow among subpopulations becomes more difficult (Bohonak 1999).
Island endemic species can also be vulnerable to invasive species (Davis 2009). For example, alien predators have greater impacts in island ecosystems because of prey naïveté, where native prey lack predator avoidance strategies (Salo et al. 2007). Serious declines have been reported for many island endemic populations that have been exposed to high predation pressures (Lewis et al. 2011; Ohbayashi et al. 2007; St Clair et al. 2011). At the same time, the high predation pressure from invasive species can cause habitat fragmentation. As an introduced species expands its habitat, prey populations can become smaller and fragmented, which might lead to deleterious effects on genetic variation. However, the impacts of invasive species on native species are largely studied in terms of ecological, rather than genetic, consequences (Gasc et al. 2010).
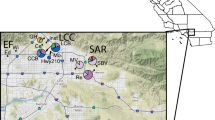
The Amami Islands, located in southern Japan, are designated as a quasi-national park due to their valuable natural environment. Several endemic species inhabit the islands, many of which are national treasures, such as the black rabbit (Pentalagus furnessi), the Amami jay (Garrulus lidthi), and the Ryukyu long-haired rat (Diplothrix legata). However, human activities such as deforestation and road construction in the forests have deteriorated the habitats of these animals (Environment Agency 2000). Furthermore, to control the native venomous pit viper, the mongoose (Herpestes javanicus), an invasive carnivore, was introduced by humans in 1979 near the centre of the Naze area on Amami-Oshima (hereafter Oshima; Fig. 1). The mongoose rapidly expanded its distribution up to 10 km from the release point by 1990, and more than 20 km by 2004 (Ministry of the Environment 2005). Rather than reducing the viper population, the mongooses prey on many native species (Abe et al. 1999) and have had strong negative impacts on rare vertebrates (Watari et al. 2008). Indeed, during a survey conducted in 2003, many native species, including the black rabbit as well as several species of frogs were not found within 10 km of the release point of the mongoose. This showed that the high predation pressure from the mongoose and the presence of the city itself, with large roads and residents, created a habitat gap around Naze area for many endemic species. Such a gap might have caused habitat fragmentation and lowered the gene flow throughout the area, thus threatening the genetic diversity of rare species on Oshima. However, studies examining the effects of the mongoose have focused only on its role as a predator, and no studies have been published on the genetic status of the endangered species on this island.

Map of the Amami Islands. The largest island is Amami-Oshima (Oshima), and the second largest one, south of and next to Oshima, is Kakeroma-jima. Black circles show the sampling localities of Babina subaspera, labelled with their three-letter codes and the number of samples in parentheses. Thick lines indicate National Road No. 58, with tunnels shown in dotted lines. Naze area is the centre of human population
The otton frog, Babina subaspera, is endemic to two of the Amami Islands, Oshima and Kakeroma-jima (Maeda and Matsui 1999). This frog has high academic value because of its unique “fifth finger” (Tokita and Iwai 2010). However, B. subaspera is now threatened due to habitat degradation and predation pressure from the mongoose, and is listed as an endangered species in the IUCN Red List of Threatened Species (IUCN 2011). A habitat gap around Naze area created by high predation pressure from the mongoose was observed for this species (Iwai and Watari 2006; Watari et al. 2008). Adult B. subaspera exhibit low dispersal abilities, often with a home range of a few 100 m during the year (Iwai, unpublished data). Therefore, habitat connectivity might be critical for maintaining its genetic diversity.
Here, we investigated the genetic diversity, differentiation and gene flow of B. subaspera populations in the Amami Islands to determine its genetic status. We used five microsatellite loci to study seven populations in Oshima surrounding the area of mongoose establishment, as well as on Kakeroma-jima. We also investigated the historical divergence within B. subaspera by examining variation in the mitochondrial genome among populations. We discuss the possible effects of invasive predators on the gene flow of native species and prospects for the conservation of B. subaspera.
Methods
Study area
Oshima and Kakeroma-jima are located in Kagoshima Prefecture, southern Japan. Both are mountainous islands covered with subtropical rain forests with the highest peak at 694 m on Oshima and 314 m on Kakeroma-jima. The two islands are 2 km apart at their closest point. Oshima, which is 712 km2 with a human population of 68,000, has several ridges and valleys with rivers (Fig. 1). Nearly 40,000 people live in Naze City, which is located on the northern part of Oshima (Centered at Naze area; Fig. 1). Because mountainous areas are not suitable for human residence, the urban areas are mostly located along the coastline near large roads. The largest road on Oshima is National Road No. 58, which crosses the island from the north through Naze area to the south (Fig. 1). With the exception of the road and associated human areas, the mountains are essentially continuous on Oshima. The mongoose was introduced to the centre of Naze area in 1979, and its population subsequently expanded toward the south-west of the island. Kakeroma-jima is only 77.4 km2 with 1,500 people and consists of continuous mountains without large rivers or roads.
Microsatellite analysis
The toes of 175 adult B. subaspera were sampled in 2004–2007 (1 in 2004, 82 in 2005, 73 in 2006, 19 in 2007) in Oshima and Kakeroma-jima. Individuals within close geographical distance were grouped into seven populations (Fig. 1). Population sample sizes ranged from 11 to 48.
DNA was extracted from ethanol-preserved toes using PrepMan Ultra Sample Preparation Reagent (Applied Biosystems). Five microsatellite loci were examined: Bas2011, Bas2037, Bas2044, Bas2051, and Bas2091 (Iwai et al. 2011). The forward primer of each primer set was end-labelled with NED (Bas2011), FAM (Bas2037, Bas2091), or HEX (Bas2044, Bas2051). Polymerase chain reaction (PCR) was performed in 20 μL of reaction mixture containing 0.7 μL of template DNA, 10 pmol of fluorescent-labelled forward primer (BEX/Japan Bio Service/Applied Biosystems) and 10 pmol of nonlabelled reverse primer (FASMAC), 200 μM each of dNTP and 1× buffer with 1.5 mM MgCl2 and 1.0 U of Ex Taq DNA polymerase (TaKaRa). GeneAmp 9700 (Applied Biosystems) was used for PCR amplifications under the following conditions: 94°C for 2 min; 35 cycles of 94°C for 30 s, 53°C for 30 s, 72°C for 45 s, and 72°C for 45 s. The amplified products were detected on an ABI 310 Genetic Analyzer with HD 400 ROX size standard (Applied Biosystems), and fragment sizes were estimated using GENESCAN 2.1 software (Applied Biosystems).
We estimated population genetic parameters for the seven populations. For analyses involving multiple comparisons, the critical probability for each test was adjusted using sequential Bonferroni correction (Rice 1989). MICROCHECKER (Oosterhout et al. 2004) was used to check microsatellite data for scoring errors and null alleles. Observed (H O) and expected heterozygosities (H E) were calculated to quantify the genetic diversity of each population. Allelic richness (A; El Mousadik and Petit 1996), F IS, and F ST were estimated and deviation from Hardy–Weinberg equilibrium (HWE) was tested by FSTAT (Goudet 1995). Tests for linkage disequilibrium were carried out with GENEPOP version 4.0.10 (Raymond and Rousset 1995). The level of population differentiation was quantified using the F ST estimator of Weir and Cockerham (1984). Evidence of recent population bottleneck was assessed for each population with BOTTLENECK v. 1. 2.02 (Cornuet and Luikart 1996) using the Wilcoxon signed-rank test. To visualise the relationships between geographical site and genetic differentiation, multidimensional scaling (MDS) was conducted on F ST/(1 − F ST), using the program SYSTAT v. 9.01 (SPSS, Inc.).
The relationship between genetic differentiation and geographical distance among pairwise comparisons of populations was tested by the Mantel test (Mantel 1967) with 9,999 randomisations using GenAlEx 6.2 (Peakall and Smouse 2006). A partial Mantel test was conducted to control for the effect of different islands (Oshima or Kakeroma-jima). Partial Mantel tests are useful for distinguishing the relationships among correlated variables. After making a predictor matrix based on islands (0 = same, 1 = different island), two matrices of the residuals of the regression between two of three variables were constructed and then compared by a standard Mantel test.
To assess the level of population structure and to assign an individual to an origin, individual-based clustering was performed using the program STRUCTURE v. 2.3.3 (Pritchard et al. 2000). Twenty runs were performed with a burn-in length of 50,000 and an MCMC of 50,000 for each K (1 to 5). As sampling location information can be used to provide accurate inferences with STRUCTURE (Hubisz et al. 2009), a LOCPRIOR model was performed. The likelihood of the assignments was evaluated for K varying from 1 to 5, and ΔK was calculated to examine the true K number (Evvano et al. 2005).
Mitochondrial DNA analysis
DNA was extracted in the same way as done for the microsatellite analysis. Fragments of 743 bp of the cytochrome c oxidase subunit I (CO I) of 23 specimens from each population (see Table 1) were amplified by PCR. The CO I is reported to exhibit high intraspecific divergence for amphibians (Vences et al. 2005). The following primers were designed for this study from the Rana holsti sequence (accession number AB511297): CO1Hol-F (forward primer 5′-CGG CCA CTT TAC CTG TGA TAT TC-3′) and CO1Hol-R (reverse primer 5′-GAC TTC AGG GTG ACC AAA GAA TC-3′). PCR was conducted under the following conditions: 94°C for 1.5 min; 35 cycles at 94°C for 30 s, 53°C for 45 s and 72°C for 1.5 min. PCR products were purified using the MultiScreen HTS™ PCR (Millipore, Billerica, MA) and sequencing was performed by SolGent Co., Ltd. (Daejeon, South Korea) using an ABI 3730xl DNA Analyser (Applied Biosystems, Foster City, CA). All fragments were sequenced in both directions. Sequences were deposited in DDBJ/EMBL-Bank/GenBank under the accession numbers AB679639–AB679645. Alignment of the CO I sequences was performed by ClustalX (Thompson et al. 1997), and the DnaSP v.4.10 (Rozas et al. 2003) software was used to estimate the nucleotide diversity (π) (Nei 1987). A minimum spanning network was constructed using the TCS software package (Clement et al. 2000), which employs a method of probability of parsimony according to Templeton et al. (1992). The maximum number of mutational steps that constitutes a parsimonious connection between two sequence types was calculated with 95% confidence.
Results
All 175 individuals from seven populations were successfully genotyped for the five loci examined. The genetic diversities within populations between different sampling years were examined in the Yuwan (YUW) and Aoku (AOK) populations, for which relatively numerous samples had been collected. Given that the pairwise F ST values between years within each population were smaller than 0.02, and the difference was not significant (adjusted P > 0.05), temporal difference had little effect on the overall results. We combined samples from the same population with different collection periods for consecutive analyses. The number of alleles varied from 14 (Bas2044) to 21 (Bas2037, Bas2091), with an average of 18 alleles per locus for all individuals. No significant linkage disequilibrium was detected in any combination of loci for the seven populations (70 tests). The mean expected heterozygosity (H E) of all loci varied from 0.666 to 0.852, and the genetic diversity, estimated as allelic richness, varied from 4.6 to 9.5 (Table 1). Heterozygote deviations from Hardy–Weinberg equilibrium were detected in the FUK and AOK populations. No recent genetic bottleneck was detected in any population (Table 1). Null alleles were suggested in Bas 2011 of TAG, Bas 2037 of YUW and AOK, Bas 2044 of AOK, Bas 2051 of KAR and Bas 2091 of FUK. Because no loci showed the possibility of null alleles in more than two populations, we considered that this was not caused by the use of inappropriate primers, and loci were used in subsequent analysis.
Pairwise F ST estimations are shown in Table 2. Significant population differentiation was detected; the TAG, AOK, and KAR populations were distinct from all other populations, whereas the values of F ST among KMY, YUW, UKN, and FUK were lower than 0.025 and were not distinct from one another, except for FUK and UKN (F ST = 0.044). Multidimensional scaling could explain 99.9% of all genetic distance among populations; the peripheral populations, TAG and KAR, were clearly divided from other midland populations KMY, YUW, UKN, FUK, and AOK, which were closely arranged (Fig. 2).

Results of the multidimensional scaling analysis based on F ST/(1 − F ST)
The Mantel test showed that between-population differentiation increased significantly with geographical distance (R 2 = 0.502, P < 0.05), suggesting a model of isolation by distance (Fig. 3). The continuous distribution but restricted dispersal of B. subaspera creates a balance between local genetic drift and geographically mediated gene flow (Wright 1943). The KAR population showed higher genetic distance than did the other 6 populations. The partial Mantel test showed that geographic distance significantly affected both KAR and other populations (R 2 = 0.544, P < 0.001), although the genetic distance was higher in KAR for the same geographic distance (R 2 = 0.600, P < 0.001).

Relationships between geographical distance and genetic differences. Open circles indicate data between Kakeroma and other populations
A substantial genetic structure was detected by STRUCTURE for all 175 individuals of B. subaspera (Fig. 4). The likelihood of assignment, lnP(D), increased from K = 1 to K = 4 and then decreased at K = 5, with a higher standard deviation (Fig. 5). ΔK was calculated for K = 1–5, and was highest at K = 2 and lowest at K = 5 (Fig. 5). Given that adding a cluster from K = 5 did not provide much more information regarding genetic structuring (Fig. 4), assignment results were interpreted for K = 2–4. When K = 2, populations were clearly divided into midland populations (KMY, YUW, UKN, FUK, and AOK) and other peripheral populations (TAG and KAR). When K = 3, TAG and KAR were each isolated from the other populations. When K = 4, midland populations were suggested to come from two different populations with some admixture. Within the midland populations, AOK seemed to have different admixture history from other midland populations (Fig. 4).

The proportion of the membership coefficient of the 175 individuals in seven populations for each of the inferred clusters for K = 2–5 in STRUCTURE analysis. Population codes are indicated between the thick vertical lines

Mean likelihood of K (left axis, ± SD, black circle) and ΔK (right axis, cross mark) for K = 1–5 in STRUCTURE analysis
We sequenced 676 bp of the mtDNA CO I region. From 23 samples only seven different haplotypes with a maximum divergence of three steps were detected (Fig. 6). The overall nucleotide diversity on the sequenced samples gave an estimated π = 0.00136 ± 0.00031. Using statistical parsimony, a single haplotype network was produced by the TCS analysis. It was characterised by a star-like topology with short branches due to a limited number of mutation steps separating the haplotypes, and one dominant central haplotype (Fig. 6). The most common haplotype was distributed on both islands and was shared by the three groups suggested by the STRUCTURE analysis.

Statistical parsimony network of the mitochondrial DNA CO I region haplotypes. The size of each circle indicates the frequency of each haplotype. A small circle with no code indicates a missing haplotype in the network that was not present in the sample. Lines connecting haplotypes represent a single mutational step regardless of the length. Site abbreviations are the same as in Table 1
Discussion
The genetic diversity of B. subaspera (H E = 0.67–0.85) was relatively high compared to that of other frog species (0.18–0.43, Allentoft et al. 2009; 0.39–0.60, Arens et al. 2006; 0.14–0.50, Blouin et al. 2010). This indicates that B. subaspera still maintains high genetic diversity even though their populations are declining. With the exception of the KAR population on Kakeroma-jima, all six populations on Oshima showed H E values higher than 0.80. No recent genetic bottleneck was detected, suggesting that the populations were not recently colonised by small numbers of individuals and have not experienced recent severe declines in population size. This also indicates that populations of B. subaspera in the Amami Islands have been well maintained to date.
According to the F ST population differentiation analyses, four major population groups of B. subaspera occur on the Amami Islands: TAG, AOK, KAR, and the remaining midland populations (except for AOK). This pattern was well supported by the results of the STRUCTURE analysis, in which these four groups showed different patterns under K = 4. However, the midland populations were not divided into any smaller subgroups when K = 3, while a higher ΔK and multidimensional scaling showed that midland populations were grouped together. This suggests that the separation of AOK from other midland populations is less complete than those of TAG and KAR from the midland populations. Thus, one may reasonably consider that B. subaspera on the Amami Islands should be divided into three main clusters: TAG, KAR and midland populations. These three clusters were not genetically differentiated on a historical timescale because our mtDNA analysis revealed that they shared common haplotypes and showed a limited number of mutation steps. The “star-like” genealogy is characteristic when all haplotypes rapidly coalesce and is a general outcome of population expansion. Thus, the observed pattern suggested by the microsatellite analysis likely reflects current gene flow in B. subaspera.
The high genetic difference of TAG and KAR from the midland populations is reasonable considering their isolation by distance: TAG and KAR are geographically far from the midland populations. However, the genetic distances of KAR from other populations were higher than those expected from the same geographical distance between populations other than KAR (Fig. 3). Furthermore, the results of the STRUCTURE analysis showed that little gene flow occurred between KAR and other populations (Fig. 4). KAR is located on Kakeroma-jima, which is divided by a 2-km channel at its nearest point to Oshima. Dispersal across this channel is likely to be difficult for B. subaspera, and the KAR population may have been prevented from outbreeding with other populations on Oshima and therefore has been genetically isolated since this geographical barrier appeared. The ancestral forms of the Amami Islands were shaped 1–1.5 MYBP (Kizaki and Oshiro 1980), and small islands have repeatedly separated and reattached since then. Oshima and Kakeroma-jima were probably separated after the last glacial period, 18,000–20,000 years ago (Matsui 1996). Given that no bottleneck was detected in KAR, the current population did not likely originate from a small number of colonisers after the geological isolation of Kakeroma-jima, but rather from those that remained on the islands when the separation occurred. These hypotheses are consistent with the observed genetic status of KAR (i.e. gene flow with midland populations once occurred and was lost relatively recently). The KAR population may have been maintained within a relatively normal population size without dispersal among the islands.
TAG was differentiated from and showed little gene flow with midland populations, although no distinct geological barrier exists between them. This isolation of TAG likely occurred relatively recently because our mtDNA analysis showed that TAG and other populations were similar on a historical timescale. Moreover, although geological barriers such as mountain ridges (Funk et al. 2005) and rivers (Simões et al. 2008) have been reported to act as a genetic barrier for amphibians, our results showed this was not the case; i.e. populations divided by ridges or rivers (e.g. between UKN and KMY) were not genetically differentiated. These data suggest that the isolation of TAG might have been caused by relatively recent, anthropogenic factors. Large roads can prevent gene flow of amphibians (Lesbarrères et al. 2006), but our results showed that the largest road in Oshima was not an effective barrier for populations (e.g. between FUK and KMY). Thus, the isolation of TAG from midland populations was likely caused by the large habitat gap between them produced by the high predation pressure from the introduced mongoose combined with the establishment of urban areas. Urban areas, however, are mostly located along the coastline or road, and do not divide mountains. Compared to the size of the habitat gap created by the mongoose (>10 km), the effect of urban area (<1 km) should be small. Thus, the introduction of this invasive predator likely resulted in the habitat fragmentation of B. subaspera, and has thereby obstructed the gene flow that previously existed between the TAG and midland populations.
Future prospects
Our study revealed that the genetic diversity of B. subaspera is still well maintained throughout the Amami Islands. Ridges, rivers or roads did not seem to prevent the gene flow of B. subaspera, suggesting that this species might have higher dispersal ability than expected. The adult B. subaspera showed a home range of only a few 100 m during a year (Iwai, unpublished data), but juveniles may disperse farther, potentially playing an important role in maintaining the apparently high genetic diversity. However, some populations exhibited lower genetic diversity and are isolated from other populations. The sea channel, as well as a large habitat gap (>10 km) appears to prevent the dispersal of B. subaspera. If the current situation does not change, the TAG and KAR populations will likely experience severe loss of genetic diversity.
With appropriate conservation measures, the gene flow between the TAG and other populations on Oshima could likely be re-established given that they are contiguous and still maintain genetic diversity. For example, the habitat gap may be overcome by the elimination of the mongoose population. Indeed, a project to eliminate the mongoose is now under way and may restore B. subaspera to the area from which it has been extirpated. The natal dispersal of B. subaspera inhabiting the nearby region should gradually fill the current gap. Of course, factors other than the mongoose, such as the existence of an urban area (<1 km wide), may also play a role in the habitat gap and may currently be masked by the strong effects of the mongoose. One must determine how these factors interact to produce the habitat fragmentation of B. subaspera. Careful monitoring of the recovery of populations and their associated genetic diversity should be conducted along as the mongoose elimination project ensues, and practical measures such as the creation of a corridor might be necessary.
The KAR population is no longer able to outbreed with other populations on Oshima. Furthermore, few appropriate B. subaspera breeding sites exist in the Kakeroma Islands (Iwai, personal observation), which may prevent B. subaspera populations from growing, resulting in a higher chance of inbreeding depression. These factors could be responsible for the observed lower genetic diversity in the KAR population, which is considered the most vulnerable population among those that we analysed. This population should be carefully maintained to prevent further loss of genetic diversity. The preservation of current breeding sites and setting limits to deforestation and the construction of roads would be beneficial.
Few studies have examined the impacts of invasive species on the genetic diversity of native species (Gasc et al. 2010). Here, we highlight the importance of evaluating whether invasive species can prevent gene flow of native species. In addition to B. subaspera, the Amami Islands are home to many endangered species, some of which have disappeared from the Naze area. To conserve these species, understanding their population genetics is essential. Along with the recovery of populations after the elimination of the mongoose, studies of these species will provide valuable information on how native species recover their genetic diversity and will serve as reference for future comparative studies.
References
Abe S, Handa Y, Abe Y, Takatsuki Y, Nigi H (1999) Food habits of Feral Mongoose (Herpestes sp.) on Amamioshima, Japan. In: Rodda GH, Sawai Y, Chiszar D, Tanaka H (eds) Problem snake management: the habu and the brown tree snake. Cornel University Press, New York, pp 372–383
Allentoft ME, Siegismund HR, Briggs L, Andersen LW (2009) Microsatellite analysis of the natterjack toad (Bufo calmita) in Denmark: populations are islands in a fragmented landscape. Conserv Genet 10:15–28
Andersen LW, Fog K, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc Lond B 271:1293–1302
Arens P, Bugter R, Westende W, Zollinger R, Stronks J, Vos CC, Smulders MJM (2006) Microsatellite variation and population structure of a recovering tree frog (Hyla arborea L.) metapopulation. Conserv Genet 7:825–834
Blouin MS, Phillipsen IC, Monsen KJ (2010) Population structure and conservation genetics of the oregon spotted frog, Rana pretiosa. Conserv Genet 11:2179–2194
Bohonak AJ (1999) Dispersal, gene flow and population structure. Q Rev Biol 74:21–25
Clement M, Posada D, Cradall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Davis MA (2009) Invasion biology. Oxford University Press, New York
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Environment Agency (2000) Threatened wildlife of Japan—red data book, 2nd edn. Japan Wildlife Research Center, Tokyo
Evvano G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Frankham R (1995) Inbreeding and extinction: a threshold effect. Conserv Biol 9:792–799
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Frankham R (1998) Inbreeding and extinction: island populations. Conserv Biol 12:665–675
Funk WC, Blouin MS, Corn PS, Maxell BA, Pilliod DS, Amish S, Allendorf FW (2005) Population structure of Columbia spotted frogs (Rana luteiventris) is strongly affected by the landscape. Mol Ecol 14:483–496
Gasc A, Durye MC, Cox RM, Kern A, Calsbeek R (2010) Invasive predators deplete genetic diversity of island lizards. PLoS ONE 5(8):e12061
Goudet J (1995) FSTAT (ver 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Hubisz M, Falush D, Stephens M, Pritchard J (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
IUCN (2011) IUCN red list of threatened species. Version 2011.1. http://www.iucnredlist.org. Accessed 10 Aug 2011
Iwai N, Watari Y (2006) Distribution of Ishikawa’s frog and the otton frog on Amami Island. Bull Herpetol Soc Jpn 2:109–114
Iwai N, Shoda-Kagaya E, Hamaguchi K (2011) Isolation and characterization of eight microsatellite loci in the Otton frog, Babina subaspera. J For Res-Jpn. doi:10.1007/s10310-011-0310-5
Kizaki K, Oshiro I (1980) The origin of the Ryukyu islands. In: Kizaki K (ed) Natural history of Ryukyus. Tsukiji-Shokan, Tokyo, pp 8–37
Lesbarrères D, Primmer CR, Lodé T, Merilä J (2006) The effects of 20 years of highway presence on the genetic structure of Rana dalmatina populations. Ecoscience 13:531–538
Lewis DS, vanVeen R, Wildon BS (2011) Conservation implications of small Indian mongoose (Herpestes auropunctatus) predation in a hotspot within a hotspot: the Hellshire Hills, Jamaica. Biol Invasions 13:25–33
Maeda N, Matsui M (1999) Frogs and toads of Japan. Revised ed. Bun-Ichi Sogo Shuppan, Tokyo
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Matsui M (1996) Natural history of the amphibia. University of Tokyo Press, Tokyo
Ministry of the Environment (2005) Annual report on eradication of introduced Javan mongoose on Amami-Oshima Island. Natural Environment Bureau, Ministry of the Environment, Tokyo
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Newman D, Pilson D (1997) Increased probability of extinction due to decreased genetic effective population size: experimental populations of Clarkia pulchell. Evolution 51:354–362
Ohbayashi T, Okochi I, Sato H, Ono T, Chiba S (2007) Rapid decline of endemic snails in the Ogasawara Islands, Western Pacific Ocean. Appl Entomol Zool 42:479–485
Oosterhout CV, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Salo P, Korpimäki E, Banks PB, Nordström M, Dickman CR (2007) Alien predators are more dangerous than native predators to prey populations. Proc R Soc B 274:1237–1243
Simões PI, Lima AP, Magnusson WE (2008) Acoustic and morphological differentiation in the frog Allobates femoralis: relationships with the upper Madeira River and other potential geological barriers. Biotropica 40:607–614
Smith FDM, May RM, Pellew R, Johnson TH, Walter KR (1993) How much do we know about the current extinction rate? Trends Ecol Evol 8:375–378
St Clair JJH, Poncet S, Sheehan DK, Szekely T, Hilton GM (2011) Responses of an island endemic invertebrate to rodent invasion and eradication. Anim Conserv 14:66–73
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data III. Cladogram estimation. Genetics 132:619–633
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tokita M, Iwai N (2010) Development of the pseudothumb in frogs. Biol Lett 6:517–520
Vences M, Thomas M, Bonett RM, Vieites DR (2005) Deciphering amphibian diversity through DNA barcoding: chances and challenges. Philos Trans R Soc B 360:1859–1868
Vitousek PM (1988) Diversity and biological invasions of oceanic islands. In: Wilson EO, Peters FM (eds) Biodiversity. National Academy Press, Washington, DC, pp 181–189
Watari Y, Takatsuki S, Miyashita T (2008) Effects of exotic mongoose (Herpestes javanicus) on the native fauna of Amami-Oshima Island, southern Japan, estimated by distribution patterns along the historical gradient of mongoose invasion. Biol Invasions 10:7–17
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1943) Isolation by distance. Genetics 28:114–138
Acknowledgments
We would like to thank Setsuko Suzuki and Noe Matsushima for their advice. We also thank Yuya Watari and Saya Uemura for their help with field collection. Samples were collected under permits nos. 204 and 703 from the Kagoshima education commission. NI was supported by a Japan Society for the Promotion of Science Research Fellowship and Research Fund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iwai, N., Shoda-Kagaya, E. Population structure of an endangered frog (Babina subaspera) endemic to the Amami Islands: possible impacts of invasive predators on gene flow. Conserv Genet 13, 717–725 (2012). https://doi.org/10.1007/s10592-012-0320-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-012-0320-7