
Abstract
Eastern hemlock (Tsuga canadensis [L.] Carr.) is an ecologically important tree species experiencing severe mortality across much of its eastern North American distribution, caused by infestation of the exotic hemlock woolly adelgid (Adelges tsugae Annand). To guide gene conservation strategies for this imperiled conifer, we conducted a range-wide genetic variation study for eastern hemlock, amplifying 13 highly polymorphic nuclear microsatellite loci in 1,180 trees across 60 populations. The results demonstrate that eastern hemlock exhibits moderate inbreeding, possibly a signature of a prehistoric decline associated with a widespread insect outbreak. Contrary to expectations, populations in formerly glaciated regions are not less genetically diverse than in the putative southern refugial region. As expected, peripheral disjunct populations are less genetically diverse than main-range populations, but some are highly genetically differentiated or contain unique alleles. Spatially explicit Bayesian clustering analyses suggest that three or four Pleistocene glacial refuges may have existed in the Southeastern United States, with a main post-glacial movement into the Northeast and the Great Lakes region. Efforts to conserve eastern hemlock genetic material should emphasize the capture of broad adaptability that occurs across the geographic range of the species, as well as genetic variability within regions with the highest allelic richness and heterozygosity, such as the Southern Appalachians and New England, and within disjunct populations that are genetically distinct. Much genetic variation exists in areas both infested and uninfested by the adelgid.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Eastern hemlock (Tsuga canadensis [L.] Carr.) is a slow-growing, long-lived conifer widely considered to be a foundation species in the forest ecosystems where it occurs (Ellison et al. 2005). It is distributed across a large geographic range extending from Nova Scotia west into Wisconsin and Minnesota and south along the Appalachian Mountains into northern Georgia and Alabama, with several peripheral disjunct populations occurring to the east and west of the main distribution (Farjon 1990). It is a wind-pollinated species that reaches reproductive maturity after two or three decades, and which typically disperses its seeds short distances (Godman and Lancaster 1990; Bonner and Karrfalt 2008). Eastern hemlock appears to be self-compatible (Bentz et al. 2002), although apparently not able to reproduce through apomixis (Nienstaedt and Kriebel 1955). It occupies multiple forest types, grows at elevations from near sea level to 1,500 m, and prefers sandy loam, loamy sand, and silt loam soils that are characteristically moist, well-drained, and acidic (Godman and Lancaster 1990). In forests where eastern hemlock is the dominant tree species, it plays a key role in stabilizing soil and maintaining water quality, and its canopies create an understory microclimate that is cool, damp, and shady and to which unique floral and faunal assemblages are adapted (Ellison et al. 2005).
Throughout much of its native range, populations of eastern hemlock are threatened by an invasive insect pest, hemlock woolly adelgid (HWA, Adelges tsugae Annand). Introduced from Japan to the eastern United States in 1951, HWA has caused widespread hemlock mortality in the Southeast, mid-Atlantic, and southern New England regions during the last two decades and may functionally eliminate eastern hemlock from the forests of eastern North America (McClure et al. 2003). Evidence suggests that hemlock extirpation from eastern forests would lead to long-term ecological consequences for forest structure and composition (Orwig and Foster 1998; Heard and Valente 2009; Spaulding and Rieske 2010), hydrological processes (Ford and Vose 2007), decomposition rates (Cobb et al. 2006; Cobb 2010), and carbon and nitrogen cycling (Jenkins et al. 1999; Nuckolls et al. 2009; Albani et al. 2010; Templer and McCann 2010). Shifts in the community structure and diversity of birds (Tingley et al. 2002; Allen et al. 2009), fish (Ross et al. 2003; Siderhurst et al. 2010), amphibians (Brooks 2001), and terrestrial and aquatic arthropods (Snyder et al. 2002; Jetton et al. 2009; Rohr et al. 2009) are also likely. The adelgid also threatens Carolina hemlock (T. caroliniana Engelm.), a rare Southern Appalachian endemic restricted to a relatively small geographic range in the Carolinas, Georgia, Tennessee, and Virginia (Humphrey 1989).
The integrated strategy to mitigate the impacts of HWA on eastern North American hemlock ecosystems includes biological control (the release of predators from the adelgid’s native range), chemical insecticides, silvicultural practices, host resistance breeding, and gene conservation (Cheah et al. 2004; Ward et al. 2004; Jetton et al. 2008b; Montgomery et al. 2009). Of these, biological and chemical controls have received the most interest, and biological control is currently considered the most promising long-term solution to adelgid management (Cheah et al. 2004). The conservation of hemlock genetic resources has also received considerable attention through a collaborative ex situ gene conservation effort between Camcore (an international tree breeding and conservation program in the Department of Forestry and Environmental Resources at North Carolina State University) and the United States Department of Agriculture Forest Service. This project involves the collection of germplasm (seeds) from populations of Carolina hemlock in the Southern Appalachian Mountains and from populations of eastern hemlock in the Southeastern, Northeastern, and Midwestern regions of the United States for long-term preservation in seed banks and the establishment of conservation seed orchards in areas that can be protected from the adelgid (Jetton et al. 2008a, b). The goal is to maintain, in perpetuity, hemlock genetic material with broad adaptability and high levels of genetic diversity that will be available for the eventual restoration of degraded or extirpated populations. Genetic diversity also provides a basis for adaptation and resilience to other sources of environmental stress and change, which is particularly important given the growing number, variety and frequency of stress exposures to tree species (Schaberg et al. 2008).
To be accomplished effectively, the conservation of eastern hemlock genetic diversity requires an understanding of range-wide population genetic structure, including the distribution of genetic variation within and among populations, the occurrence of rare alleles, and levels of inbreeding (Eriksson et al. 1993). Eastern hemlock has demonstrated morphological variation over both large and small scales, including clinal variation in seedling growth responses to photoperiod and chilling (Olson and Nienstaedt 1957; Nienstaedt and Olson 1961), the existence of two physiological races in Wisconsin (Eickmeier et al. 1975), and the widespread co-occurrence of two types distinct in terms of growth rate, morphology and response to macroclimatic variation (Kessell 1979). A series of regional studies have assessed population-level genetic variation in eastern hemlock using biochemical and DNA markers. An analysis of highly conserved chloroplast DNA polymorphisms primarily among disjunct populations identified little differentiation (Wang et al. 1997), while Zabinski (1992) detected little allozyme variation within eastern hemlock in a study that was focused in the Midwestern portion of the eastern hemlock range. In a study that sampled 20 populations from the Southeastern portion of the range, Potter et al. (2008) also found low allozyme diversity in hemlock but reasonably high population differentiation. A range-wide analysis using seven haploid chloroplast DNA loci (Lemieux et al. 2011) also detected low among-population differentiation and, consistent with Potter et al. (2008), greater differentiation and relatively high allelic richness among southeastern Appalachian populations. These results led both sets of authors to conclude that the majority of eastern hemlock genetic diversity may have originated from a Pleistocene glacial refuge in this region. Further screening from nuclear or mitochondrial DNA markers is necessary, however, to strengthen inferences regarding the distribution of the species during the last glacial maximum (Lemieux et al. 2011). A more robust analysis of genetic variation across the range of the species also is needed to improve the efficiency of existing ex situ and in situ conservation efforts given eastern hemlock’s large natural distribution, the existence of disjunct populations that may possess unique adaptive characteristics, and the fact that HWA has already infested and significantly impacted approximately 50% of hemlock ecosystems in the eastern United States (United States Department of Agriculture Forest Service 2011).
We used 13 highly polymorphic microsatellite molecular markers to assess genetic structure and variation across the geographic range of eastern hemlock. We amplified microsatellite marker loci isolated from eastern and Carolina hemlock (Josserand et al. 2008; Shamblin et al. 2008) from 1,180 trees in 60 populations to conduct a range-wide assessment of eastern hemlock genetic diversity and to (1) identify areas of high and low genetic variation, (2) evaluate genetic variation in peripheral disjunct and main range populations, (3) assess regional differences in genetic variation to better understand the recent phylogeographical history of the species, and (4) compare genetic variation in populations currently threatened by HWA with those not yet within the generally infested range.
Materials and methods
Sample collection and DNA extraction
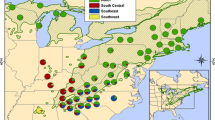
This analysis encompassed foliage samples from a total of 1,180 trees representing 60 populations from across the entire range of eastern hemlock (Table 1; Fig. 1). Additionally, 10 samples of Carolina hemlock from Craig County, Virginia, were included as an outgroup population. Of the eastern hemlock samples, 400 were collected during February and March of 2006 from 20 populations in the southern portion of the range. In 2009, samples were collected from an additional 780 trees from 32 locations from across the northern part of the range and from eight disjunct populations in the South. Populations were defined as northern or southern based on their location relative to the maximum extent of the Wisconsinian glaciation ca. 18,000 years before present (Dyke et al. 2003) (Fig. 1). In nearly all populations, samples were collected from 20 trees spaced at least 100 m apart. This strategy, which is consistent with established gene conservation strategies employed in eastern hemlock seed collections, diversifies population-level seed collections by avoiding the sampling of neighbors that may be related as a result of short-distance seed or pollen dispersal (Brown and Hardner 2000). Such short-distance gene dispersal is probably common in eastern hemlock because most seeds fall within tree height because of their small wings (Godman and Lancaster 1990). Additionally, despite being dispersed by wind (Godman and Lancaster 1990), eastern hemlock pollen is poorly dispersed (Jackson 1991).

Sampled populations of eastern hemlocks (Tsuga canadensis). See Table 1 for population information. Populations identified as existing within counties where hemlock woolly adelgid was absent or present in 2010 (United States Department of Agriculture Forest Service 2011). Maximum extent of the Wisconsinian glaciations, ca. 18,000 years before present, is from Dyke et al. (2003)
Populations were classified as isolated disjuncts if they are located more than 10 km outside the edge of continuous main range of the eastern hemlock distribution as defined by Little (1971). These seven disjunct populations were an average of approximately 147 km from the main eastern hemlock range (farthest: Shades State Park, ca. 315 km; nearest, James River, ca. 22 km). Several populations were nearer to other disjuncts than to the main range, but most were still highly isolated (Bankhead National Forest, ca. 110 km; Shades State Park, ca. 64 km; Hemlock Cliffs, ca. 56 km; Mammoth Cave National Park, ca. 71 km). The mean distance to the nearest eastern hemlock population, main range or disjunct, was ca. 68 km. Hemlock Bluffs was the most isolated population overall (ca. 140 km). Populations were characterized as threatened by hemlock woolly adelgid based on the presence of established HWA infestations within their counties as of 2010 (United States Department of Agriculture Forest Service 2011).
All 1,180 foliage samples were kept in cold storage until sent to the National Forest Genetics Laboratory (NFGEL) in Placerville, California, for DNA extraction and microsatellite analysis. Needles from the 400 samples collected in 2006 were also used in an allozyme analysis of southern hemlock populations (Potter et al. 2008). Genomic DNA for all samples was extracted from the needle samples using the DNEasy 96 Plant Kit (Qiagen, Chatsworth, California, USA). DNA concentrations were determined using a Gemini XPS Microplate Spectrofluorometer (Molecular Devices, Sunnyvale, California, USA) with PicoGreen dsReagent (Invitrogen, Carlsbad, California, USA).
Microsatellite analysis
To select a set of microsatellite markers for this study, we evaluated a total of 42 microsatellite primer pairs at NFGEL for usefulness in examining eastern hemlock genetic structure and diversity. Of these, 29 primer pairs had been isolated from eastern hemlock (Shamblin et al. 2008), six had been isolated from Carolina hemlock and had previously demonstrated amplification and polymorphism in eastern hemlock (Josserand et al. 2008), and seven had been isolated from western hemlock (Tsuga heterophylla (Raf.) Sargent) (Amarasinghe et al. 2002; Wellman et al. 2003). Initial screening and optimization was undertaken on genomic DNA extracted from a subset of eastern hemlock samples collected in 2006. Primer pairs that generated polymorphic, easily identifiable and consistently amplified fragments were then run across the 400 samples collected in 2006. The genotyped data from each locus were tested for high null allele presence (Brookfield 1996) using Micro-Checker 2.2.3 (van Oosterhout et al. 2004), and those with low estimated proportions of null alleles (<0.1) were subsequently run across the rest of the 1,180-sample pool.
We genotyped nine promising eastern hemlock primer pairs across the Southern samples, and we selected eight for inclusion in the full population genetic analyses (Table 2). We selected none of the seven western hemlock primer pairs for inclusion in analysis, although we ran and genotyped one (EE10) for all the 2006 samples but subsequently discarded it because of high null allele frequency. We selected five Carolina hemlock primer pairs for inclusion in the study (Table 2). For the Carolina hemlock-derived loci, polymerase chain reaction (PCR) amplification was performed in 15-μl reaction volumes containing 20 ng genomic DNA, 0.04 μM of the M13-tailed forward primer, 0.16 μM of the dye-labeled M13 universal primer, 0.16 μM unlabeled and untagged reverse primer, 167 μM each dNTP, 1× Taq buffer, 3.17 mM MgCl2, and 0.2 units of HotStarTaq DNA polymerase (Qiagen, Valencia, CA) for all markers with the exception of TcSI_083, for which 0.3 units were used. Additionally, 0.67 mg/ml of bovine serum albumin (BSA) were added for TcSI_029 and TcSI_052. The PCRs were completed with the following protocol on PTC-100 thermal cyclers (MJ Research, Watertown, MA): 15 min at 95°C; 20 cycles of 30 s at 94°C (denaturation), 30 s at 65°C for the first cycle minus 0.5°C each subsequent cycle (annealing), and 1 min at 72°C (extension); and 25 cycles of 30 s at 92°C, 30 s at 55°C, and 1 min 30 s at 72°C; all followed by a final 15 min extension at 72°C and an indefinite hold at 4°C.
For eastern hemlock-derived loci, PCR amplification was performed in 10-μl reaction volumes containing 20 ng genomic DNA, 167 μM each dNTP, 1× Taq buffer, and 0.5 units of HotStarTaq DNA polymerase. In addition, 3.0 mM MgCl2 was added for Tcn3H04, Tcn10A07, Tcn10B01, and Tcn10D07. For two loci, Tcn7H12 and Tcn10D07, reactions were run with 0.5 μM of a labeled forward primer and 0.5 μM of an unlabeled reverse primer. For the rest, reaction mixtures contained 0.05 μM of the M13-tailed forward primer, 0.45 μM of the dye-labeled M13 universal primer, 0.5 μM unlabeled and untagged reverse primer. The PCR protocol for the eastern hemlock loci was 15 min at 95°C; 21 cycles of 20 s at 95°C (denaturation), 30 s at 65°C for the first cycle minus 0.5°C each subsequent cycle (annealing), and 1 min at 72°C (extension); and X cycles of 20 s at 95°C, 30 s at 54.5°C, and 1 min at 70°C (where X was 15 for Tcn7H12 and Tcn10B01, 24 for Tcn2C05 and Tcn10D07, and 34 for the rest of the eastern hemlock markers); all followed by a final 10 min extension at 72°C and an indefinite hold at 4°C.
The PCR products for all markers were separated on an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA), as recommended by the manufacturer. Peaks were sized and binned, and then alleles were called using GeneMarker 1.51 (SoftGenetics, State College, PA), with GS(500-250)LIZ as an internal size standard for each sample. Visual checks were performed on all peaks.
Bayesian assignment tests
Large-scale migration and admixture of distinct gene pools within species may result from complex spatiotemporal processes within species (Durand et al. 2009), such as the responses to the Quaternary ice ages that are thought to have played an especially important role in determining the current genetic structure of species and populations (Hewitt 2000). Bayesian clustering models that explicitly include geographical information in the inference of population structure can prove helpful in the detection of admixture in secondary contact zones (Durand et al. 2009), such as those created by the migration and contact of gene pools from separate Pleistocene glacial refuges. To infer the number and composition of genetic clusters of eastern hemlock, we used two model-based Bayesian clustering approaches that analyzed the microsatellite genotypes for each individual tree, TESS 2.3.1 (Chen et al. 2007) and STRUCTURE 2.3.3 (Pritchard et al. 2000).
In TESS, we first used a non-admixture analysis to narrow the range of the maximum possible clusters, using a burn-in period of 10,000 replicates and 50,000 total Markov Chain Monte Carlo iterations, as recommended by Durand et al. (2009). We conducted 10 runs for each number of possible maximum clusters (K) from 2 to 12, then calculated and plotted the mean deviance information criterion (DIC) values for each K. Using this statistical model selection approach facilitated by TESS, which computes DIC as the average deviance over a run plus a penalty for the number of model parameters (Durand et al. 2009), we selected 8 as the upper bound of K for the subsequent admixture analysis, based on where the DIC values reached a plateau. Admixture models are more flexible and more robust than models without admixture (Francois and Durand 2010); the hierarchical Bayesian algorithm implemented in TESS includes the ability to include spatial prior distributions on individual admixture proportions. We used the conditional autoregressive (CAR) Gaussian model with a quadratic trend degree of two (Besag 1975; Durand et al. 2009) to estimate admixture proportions from K = 2 to K = 8, with 10 runs for each K, with 20,000 burn-in replicates and 70,000 total sweeps. The plotted mean DIC values for each K showed a plateau beginning at K = 4. We therefore implemented 100 more admixture runs in TESS with K = 4, again with 70,000 total MCMC iterations (20,000 burn-in), and selecting the top 10% based on the highest likelihood DIC values.
In STRUCTURE, we conducted an admixture analysis, assuming uncorrelated allele frequencies, with 20,000 burn-in replicates and 70,000 total sweeps. We used sample locations as prior information to assist the clustering, which allows genetic structure to be detected at lower levels of divergence or with less data than the original STRUCTURE models, and is therefore not biased towards detecting structure that is not present (Hubisz et al. 2009). We ran the model 20 times for each K from 1 to 12. While the log-likelihood values of K peaked at 4, the ΔK statistic of Evanno et al. (2005) peaked at K = 3, suggesting this is the most likely number of genetic clusters.
We exported the results from the final TESS and STRUCTURE cluster analyses to CLUMPP version 1.1.2 (Jakobsson and Rosenberg 2007) to generate an averaged Q matrix of individual posterior cluster probabilities, using the greedy algorithm and the G’ pairwise matrix similarity statistic. We then calculated the proportion of overall genetic cluster presence probability for each of the 60 eastern hemlock populations, based on the probability of cluster membership for the individuals in the population. We displayed these population-level probability proportions in map form using ArcMap 9.2 (ESRI 2006).
TESS has performed well compared to other programs in correctly estimating numbers of populations (Chen et al. 2007; Francois and Durand 2010) and has proved successful in detecting admixture in secondary contact zones, both providing a correct description of smooth clinal variation and detecting zones of sharp variation present in the data (Durand et al. 2009). Additionally, the spatially explicit nature of the TESS algorithm may be more likely to correctly reflect weak but significant patterns of isolation by distance detected in our data (see “Results”). We therefore conducted further analyses based on the results of the TESS-inferred clusters, assigning each tree to the TESS-inferred genetic cluster to which it had the highest probability of belonging.
Genetic variation and differentiation analyses
Allele calls from the 13 microsatellite loci were used to conduct analyses of genetic variation across loci and at the population level, and to conduct analyses of genetic differentiation for the Bayesian-inferred clusters. We used GENEPOP 4.0.10 (Raymond and Rousset 1995) to conduct Fisher’s exact tests for Hardy–Weinberg equilibrium for each locus and population, with 100 batches and 1,000 iterations, then used the MULTTEST procedure in SAS 9.2 (SAS Institute Inc. 2008) to calculate q values (p values adjusted for the false discovery rate associated with multiple comparisons). We used FSTAT, version 2.9.3.2 (Goudet 1995) to test for linkage disequilibrium between pairs of loci, based on 1,560 permutations and adjusted for multiple comparisons. We estimated null allele frequencies across the entire sample pool using Micro-Checker 2.2.3 (van Oosterhout et al. 2004).
We used GENEPOP to estimate inter-population gene flow (Nm) among TESS-inferred genetic clusters under the private allele method (Barton and Slatkin 1986), corrected for sample size. We used FSTAT version 2.9.3.2 (Goudet 1995) to calculate allelic richness (A) and Weir and Cockerham’s (1984) within-population inbreeding coefficient population inbreeding coefficient (F IS ) values across loci. We separately used FSTAT allele frequency outputs to determine the number of unique (private) alleles per population (A U ). We also used FSTAT to estimate among-population F ST values across eastern hemlock, and pairwise F ST values between populations and between the genetic clusters inferred in TESS. Using the program SMOGD (Crawford 2010), we calculated per-locus estimates of Jost’s D (Jost 2008), D est , as a measure of genetic differentiation across all populations of eastern hemlock, and as a measure of differentiation between pairs of clusters. Jost’s D is considered a more mathematically consistent description of population structure than the widely calculated F ST and its relatives (Jost 2008); we include both D est and F ST for comparison. We then calculated the arithmetic means of D est across the loci for eastern hemlock, and calculated the 95% confidence interval for this mean using the confidence intervals of each locus. We calculated expected heterozygosity (H E ) with Arlequin 3.0 (Excoffier et al. 2005) for each locus and population, and then used the values of H E to calculate the effective number of alleles A E as 1/(1 − H E ) (Jost 2008). We also used outputs from Arlequin to calculate mean observed heterozygosity (H O ) across all loci and percent polymorphic loci (Pp) for each population.
To assess whether eastern hemlock or any of the sampled populations or inferred genetic clusters had experienced population bottlenecks in the recent past, we used the software package Bottleneck 1.2.02 (Piry et al. 1999) to compute the difference, averaged over loci, between actual heterozygosity and the heterozygosity that would be expected if the population were in mutation-drift equilibrium. An excess of heterozygosity is expected to be consistent with a recent population bottleneck, while a deficiency of heterozygotes suggests recent population expansion without immigration (Cornuet and Luikart 1996; Karhu et al. 2006). We used a two-phase model (TPM) of microsatellite mutation, which is an intermediate between the single mutation model (Kimura and Ohta 1978) and the infinite alleles model (Kimura and Crow 1964). In keeping with the presumed model for microsatellites (Piry et al. 1999), the parameter settings consisted of 95% one-step mutations (95%) and 5% multiple-step changes, with 12% variance in multistep mutations. Significance of heterozygosity excess or deficiency was evaluated with a one-sided Wilcoxon sign-rank test using 5,000 simulation iterations. Since no populations exhibited heterozygosity excess, we reported p values from tests of heterozygosity deficiency (H def ). Additionally, we tested whether populations exhibit an L-shaped distribution of alleles (many low-frequency alleles and a few high-frequency alleles); a mode shift from this distribution is expected when a population has experienced a bottleneck (Luikart et al. 1998).
To visualize potential evolutionary relationships among the TESS-inferred genetic clusters, we constructed a neighbor-joining (NJ) (Saitou and Nei 1987) phylogram using the SEQBOOT, GENDIST, NEIGHBOR, and CONSENSE components of PHYLIP 3.6 (Felsenstein 2005). The NJ algorithm is a robust method for constructing trees from genetic distances (Mihaescu et al. 2009). The phylogram was computed from cluster allelic frequencies using chord genetic distance (D C ) (Cavalli-Sforza and Edwards 1967), which does not require assumptions about the model under which microsatellites mutate and is considered superior to most others in phylogenetic tree topology construction over short spans of evolutionary time (Takezaki and Nei 1996; Libiger et al. 2009). Confidence estimates associated with the topology of the NJ phylogram were determined with 1,000 bootstrap replicates. Carolina hemlock was included as an outgroup. We also used the GENDIST component of PHYLIP 3.6 (Felsenstein 2005) to determine pairwise D C distances among all 60 eastern hemlock populations, which we then used to calculate mean D C between each population and every other population as a measure of overall genetic distance.
We tested for isolation by distance (IBD) by conducting a Mantel test for correlations between matrices of pairwise interpopulation geographic distances and pairwise interpopulation D C genetic distances, using 9,999 permutations in GenAlEx 6.4 (Peakall and Smouse 2006). We conducted these Mantel tests separately for three groups of populations: for all populations, for northern populations, and for southern populations.
Comparisons of groups by geography, infestation and isolation
Using the UNIVARIATE procedure in SAS 9.2 (SAS Institute Inc. 2008), we calculated within-group population means for several genetic variation metrics (A, A U , P P , H O , H E , A E , F IS , mean pairwise D C with all other populations, and H def p value), for populations north and south of the maximum glacial extent, for populations in or not in counties currently infested with hemlock woolly adelgid, and for populations within and disjunct from the main range of the eastern hemlock distribution. To test the null hypothesis that there was no significant difference between the means of each pair of groups (north vs. south, inside vs. outside infested counties, interior vs. disjunct), we conducted an exact two-sample Wilcoxon rank-sum test using the NPAR1WAY procedure in SAS, with 10,000 Monte Carlo runs generating p values, then employed the MULTTEST procedure to calculate q values. Finally, we used the CORR procedure in SAS 9.2 to test for Pearson correlations between the genetic variation metrics and population latitude, longitude, and elevation.
Results
Bayesian cluster assignment
The spatially explicit Bayesian clustering analysis of individual trees using TESS 2.3.1 inferred the existence of four genetic clusters in eastern hemlock (Fig. 3a). This approach found only one of these clusters to be common in the northern populations, while all four were inferred in the southern part of the range. The North cluster was common in populations along the crest of the Southern Appalachians, but was relatively uncommon in, or absent from, populations to the west of this mountain chain. The second most prevalent genetic cluster, in the South Central portion of the species distribution, was relatively common in the Southern Appalachians, decreasing in importance among populations in the Central Appalachian highlands and Mid-Atlantic states. It was most common in populations to the west of the Southern Appalachians, accounting for the majority of the genetic makeup of populations in the Cumberland Plateau region of Tennessee, and in Kentucky and Indiana. It was also important in the two Ohio populations. A Southwest genetic cluster, meanwhile, predominated only in the isolated disjunct Bankhead National Forest population in Alabama. A Southeast genetic cluster made up the majority of the genetic composition of the Hemlock Bluffs isolated population in North Carolina, was common in Southern Appalachian populations, and was present in small proportions in the Central Appalachians.
STRUCTURE 2.3.3, meanwhile, inferred the existence of three genetic clusters. As with the TESS analysis, a single cluster was inferred as predominating in the northern parts of the hemlock range, while all clusters were present in the southern portion (Fig. 3b). A Southwest cluster was, again, associated almost entirely with the Bankhead National Forest population in Alabama, while a South Central cluster was again highly important in the other populations west of the Southern Appalachians. It was also relatively common in the Southern Appalachians and in the isolated Hemlock Bluffs population of North Carolina, appearing to take the place of the Southeast genetic cluster inferred by TESS.
For further analysis of broad-scale evolutionary patterns, each tree was assigned to the TESS-inferred genetic cluster to which it had the highest probability of belonging (n = 906 for the North cluster; n = 20 for Southwest; n = 186 for South Central; n = 68 for Southeast). A consensus neighbor-joining phylogram of D C genetic distance among these genetic clusters showed high bootstrap support (94.5%) for the grouping of the North, Southeast and South Central genetic clusters (Fig. 4). The Southwest genetic cluster was external to this clade. Within the clade, the North and Southeast clade grouped with moderate support (45% of bootstrap replicates).
Pairwise comparisons of migration between genetic clusters (Nm) suggested a high level of historical gene flow between the North and South Central clusters: 12.81 migrants per generation, corrected for sample size (Table 4). Inferred gene exchange between any other pair of clusters, however, was generally low, with the lowest values associated with the isolated Southwest cluster. Pairwise estimates of between-cluster differentiation again indicated a close genetic relationship among the North, South Central and Southeast genetic clusters (Table 4). The Southwest cluster, meanwhile, was highly differentiated from all other clusters.
Population-level genetic variation and differentiation
The 13 microsatellite loci included in this study averaged 11.23 alleles per locus across the 1,180 in-group samples (Table 2), ranging from a minimum of six alleles (Tc7H12 and Tc10A07) to a maximum of 18 (Tc10D07) at a single locus. Expected heterozygosity was moderate (mean of 0.609 across loci), but exact tests for Hardy–Weinberg equilibrium indicated a significant deficit of heterozygotes for all but two of the loci (Tc7H12 and TcSI_029). Observed heterozygosity (mean 0.529) was markedly lower than expected heterozygosity across most loci (mean 0.609). The significantly positive inbreeding coefficient (F IS ) of 0.071 (95% confidence interval: 0.037–0.108) was indicative of a deficit of heterozygotes and the likely presence of inbreeding. No linkage disequilibrium was apparent between any pairs of loci after adjusting the p value for multiple comparisons. The estimated proportion of null alleles was low (<0.1) for all 13 loci, with eight ≤0.05 (Table 2).
Estimates of among-population differentiation using Jost’s D (Jost 2008), D est , and the traditional F ST (Weir and Cockerham 1984) were divergent (Table 2). The F ST analysis estimated a moderate amount of genetic differentiation among rather than within populations (6.5%, mean F ST across loci = 0.077, 95% confidence interval: 0.055–0.078). D est was higher, with a mean across loci of 0.134 (95% confidence interval: 0.122–0.146). Estimated inter-population gene flow using the private allele method (Nm) was estimated at 6.34 migrants per generation.
The 60 eastern hemlock populations averaged 4.9 alleles per locus (A) and 2.33 effective alleles per locus (A E ) (Table 3). In general, populations with the greatest allelic richness (A) were located in the Southern Appalachians and in New England and New York (Fig. 2a). The Carolina Hemlock Campground population in North Carolina had the most alleles per locus (6.00), followed by Caesars Head State Park in South Carolina and Mount Riga State Park in Connecticut (5.69 each) and Cradle of Forestry in North Carolina and Caywood Point in New York (5.62 each). Two isolated disjuncts had the lowest allelic richness: Hemlock Bluffs Nature Preserve population in North Carolina (2.00) and Hemlock Cliffs in Indiana (2.62) (Table 3). Isolated populations across the range of the species had markedly lower allelic richness (Fig. 2a). For most populations, 100% of the loci were polymorphic (mean 97.18), with only three populations possessing fewer than 12 out of 13 polymorphic loci: Hemlock Bluffs (9 loci), Hemlock Cliffs (11 loci) and James River (11 loci) (Table 3). It is worth noting, however, that the Hemlock Bluffs and James River samples encompassed fewer than 20 trees (5 and 15, respectively) because of relatively small population sizes and our limitation on distances between sampled trees.

Eastern hemlock population classifications of a alleles per locus (A), b unique alleles (A U ), c inbreeding coefficient (F IS ), and d mean pairwise chord distance (D C ), based on 13 polymorphic nuclear microsatellite loci
Populations containing unique (private) alleles occurred throughout the range of eastern hemlock, but the populations with the most such alleles were located near the western edge of the species distribution (Table 3; Fig. 2b): Cross Village in Michigan and Shades State Park in Indiana (both with three), and Mohican-Memorial State Forest in Ohio (with two). This pattern may result in part from a lower sampling intensity in the northwestern part of the species’ range. Three disjunct populations, in addition to Shades State Park, contained a single unique allele: Bankhead National Forest in Alabama, Hemlock Cliffs in Indiana, and Quantico in Virginia (Fig. 2b).
The mean expected heterozygosity across populations (0.566) was higher than the mean observed heterozygosity (0.526) (Table 3). In general, higher values of H O and H E tended to occur in populations in the Southern Appalachians and in scattered locations across the North. Isolated disjunct populations again had the lowest values of H O and H E : Hemlock Cliffs (0.312 and 0.329, respectively), Hemlock Bluffs (0.323 and 0.350), James River (0.359 and 0.468) and Quantico (0.363 and 0.491). Twenty-nine of the 60 populations were significantly out of Hardy–Weinberg equilibrium, and the mean F IS inbreeding coefficient was 0.073 across the populations, 50 of which had positive F IS values (Table 3), all suggesting widespread inbreeding. While the Providence population in Rhode Island was most inbred (0.266), the most highly inbred populations tended to be located in the southern part of the species range (Fig. 2c). The least inbred populations were the Penobscot Experimental Forest in Maine (−0.152) and Point Beach State Forest in Wisconsin (−0.117) (Table 3).
Eastern hemlock as a whole did not exhibit the excess of heterozygosity, relative to the heterozygosity expected with mutation-drift equilibrium, that is expected following a recent genetic bottleneck. In fact, we found the opposite: significant heterozygosity deficiency (p = 0.0006), suggesting a relatively recent population expansion without immigration (Cornuet and Luikart 1996; Karhu et al. 2006). Additionally, no individual populations exhibited a significant excess of heterozygosity, but 12 had a significant deficiency at α = 0.05, nine in the north and three in the south (Table 3). Three populations did, however, possess the allele distribution mode shift expected to accompany a bottleneck: Hemlock Bluffs and South Mountains State Park in North Carolina, and Sanders Preserve in New York. Among the four TESS-inferred genetic clusters, the widespread North and South Central clusters had significant heterozygosity deficiency (p = 0.0002 and 0.0003, respectively), while the more limited Southwest and Southeast clusters did not exhibit significant excess or deficiency (p = 0.2349 and 0.1879, respectively, for heterozygosity deficiency).
Isolated disjunct populations of eastern hemlock appeared to be among the most genetically distinct based on mean pairwise chord genetic distance (D C ) between a given population and the 59 others (Table 3; Fig. 2d). These include Hemlock Bluffs (mean D C = 0.154), Bankhead National Forest (0.135), Hemlock Cliffs (0.095), Shades State Park (0.078), James River (0.064), and Mammoth Cave National Park (0.058). Populations with the lowest level of differentiation tended to exist in the Northeastern United States (Fig. 2d). Population pairwise D C and F ST values are provided in Online Resource 1 and 2.
Mantel tests revealed no evidence of isolation by distance across all 60 populations in the study (r = 0.083, p = 0.139), but did find moderate IBD among northern populations (r = 0.373, p = 0.0004) and southern populations (r = 0.390, p = 0.002).
Group comparisons by geography, infestation and isolation
Standard measures of genetic variation were not significantly different between populations north and south of the maximum extent of the Wisconsin glaciation (Table 5a). Southern populations, however, were significantly more genetically differentiated (mean pairwise D C = 0.054 vs. 0.041, p < 0.001, q = 0.002) and more inbred (mean F IS = 0.092 vs. 0.052, p = 0.04), although the inbreeding difference was not significant when accounting for the false discovery rate associated with multiple comparisons (q = 0.113). The probability of heterozogosity deficiency was significantly higher (lower H def p value) for northern populations than for southern populations (H def = 0.158 vs. 0.353, p < 0.001, q = 0.002), suggesting that northern populations are, on average, more likely to have undergone a relatively recent expansion. Few significant differences also existed between populations in counties that had and had not yet been infested by hemlock woolly adelgid (Table 5b). Populations in uninfested counties on average had more unique alleles (0.682 vs. 0.184, p = 0.011), although this difference was not significant when accounting for the false discovery rate (q = 0.103). The probability of heterozygosity deficiency also was higher among populations in uninfested counties (0.204 vs. 0.291, p = 0.041), but, again, the difference was not significant when accounting for the false discovery rate (q = 0.186). At the same time, we detected several significant differences between disjunct and interior populations (Table 5c). Specifically, interior populations were significantly more genetically diverse than isolated disjunct populations, on average, for most standard measures, even when applying more conservative q value. Inbreeding was an exception, with no significant difference between interior and disjunct populations. Disjunct populations were more highly differentiated than interior populations, according to mean pairwise D C between a given population and all other populations (0.086 vs. 0.042, p < 0.001, q < 0.001). The probability of a recent population expansion (H def p value) was higher in non-disjunct populations, but the difference was not significant when considering the false discovery rate (H def = 0.238 vs. 0.414, p = 0.049, q = 0.055).
Finally, we detected correlations each between population latitude, longitude and elevation, and some genetic diversity and differentiation measures (Table 6). We found moderate negative correlations between latitude and inbreeding coefficient (r = −0.254, p = 0.05, q = 0.134), suggesting that more northerly populations are less inbred, and between latitude and mean pairwise D C (r = −0.340, p = 0.008, q = 0.063), suggesting that more northerly populations are less genetically differentiated. Finally, we detected a moderate negative correlation between latitude and probability of heterozygosity deficiency, indicating that more northerly populations are more likely to have undergone recent expansion (r = −0.282, p = 0.029, q = 0.116). None of these relationships was significant at α = 0.05 when applying the more conservative q value, however. Longitude, having increasingly negative values for populations farther west (Table 1), was also negatively correlated with genetic distance (r = −0.278) and probability of heterozygosity deficiency (r = −0.263), indicating greater genetic differentiation for more westerly populations and greater likelihood of recent expansion among more easterly populations. Again, however, these correlations were not significant at α = 0.05 for the more conservative q value. Population elevation was positively correlated with allelic richness (r = 0.383), observed heterozygosity (r = 0.261), expected heterozygosity (r = 0.380), and effective alleles (r = 0.410); all these relationships, with the exception of elevation and observed heterozygosity, were significant at α = 0.05 for both p and q.
Discussion
Strong and sometimes unexpected geographic patterns of genetic variation exist across the eastern hemlock range. Specifically, this study appears to detect the genetic signatures of Pleistocene glacial refuges and post-glacial colonization routes of eastern hemlock, and of a relatively recent population expansion. The results also establish the existence of a negative relationship between population isolation and genetic diversity and a positive relationship between population isolation and genetic differentiation. These findings have profound gene conservation implications for eastern hemlock.
The distribution of temperate species and their patterns of genetic diversity have been shaped in large part by the periodic glacial episodes of the late Quaternary period, during which ice sheets advanced and retreated on a 100,000-year cycle. Species generally survived glacial maxima by retreating to refuges at lower latitudes (Hewitt 1996, 2000; Provan and Bennett 2008). Molecular marker studies have been widely employed to infer the location of these glacial refuges and the routes of post-glacial colonization routes for tree species (e.g., Petit et al. 2002; Heuertz et al. 2004; McLachlan et al. 2005; O’Connell et al. 2008).
Two main genetic diversity patterns support the hypothesis that a species has undergone range contraction and expansion coinciding with glacial maxima and minima (Provan and Bennett 2008): (1) Populations in areas of glacial refuges are expected to harbor higher levels of genetic diversity than areas colonized from these refuges (Comes and Kadereit 1998; Taberlet et al. 1998). This is the “southern richness and northern purity” scenario that occurs when recolonizing populations descend from subsets of the genotypes present in the refugial population and often subsequently undergo founder effects and genetic bottlenecks (Hewitt 1999). As a result, stable “rear edge” populations are of critical conservation importance as long-term collections of genetic variation and as hot spots of speciation (Hampe and Petit 2005). (2) Distinct genetic lineages are expected to exhibit spatial structuring, both between refugial areas and along recolonization routes (Hewitt 1996), because the long-term isolation of populations within geographically distinct refuges will lead to genetic differentiation and drift (Provan and Bennett 2008).
The existence of several distinct genetic clusters in eastern hemlock, as inferred by two different spatial Bayesian clustering approaches, is consistent with this second key indicator of range contraction and expansion. Specifically, this result suggests that the species was confined to three or four separate glacial refuges in the Southeastern United States. The closely related North and South Central genetic clusters (Fig. 3; Table 4), which according to both clustering approaches account for most of the genetic composition of the sampled populations, may have descended from refuges located in the vicinity of the Southern Appalachian Mountains. Higher levels of allozyme and chloroplast DNA variation in populations in the southeastern Appalachians suggest that at least one glacial refuge area may have been located in or near this region (Potter et al. 2008; Lemieux et al. 2011). Such a pattern, where most of a species’ existing distribution descends from a subset of the putative glacial refuges, is common among taxa that increased their ranges and abundances during the glacial-interglacial transition (Bennett and Provan 2008). The North genetic cluster descendants of these refugial trees may have migrated northeast along the Appalachians into the northeastern United States and southeastern Canada before colonizing the Great Lakes region. It perhaps made secondary contact with trees from the South Central genetic cluster, with which it appears to have had high levels of historical gene exchange (Table 4), consistent with chloroplast DNA evidence of homogenizing gene flow among eastern hemlock populations during the Holocene (Lemieux et al. 2011). Trees from the South Central genetic cluster, meanwhile, also may have made their way up the Appalachians in addition to moving into the Midwest. The Southwest genetic cluster, according to both clustering approaches, exists almost entirely within the Bankhead National Forest population, part of an area of remnant hemlock on the Appalachian Plateau of Alabama, where the species has perhaps remained throughout much of the Holocene (Hart and Shankman 2005). The Southeast genetic cluster inferred by TESS, meanwhile, consists of the very small Hemlock Bluffs outlier population in central North Carolina (Holmes 1883; Oosting and Hess 1956) along with portions of populations in the Southern Appalachians. Perhaps this genetic cluster descends from the remnant hemlock that disappeared from the coastal plain of the central Atlantic Seaboard between 4,000 and 2,000 years ago (Delcourt and Delcourt 1987). On the other hand, the Southeast, North and South Central clusters are all closely related (Table 4; Fig. 4) and STRUCTURE did not infer the existence of the Southeast cluster, suggesting that these three clusters may have resulted from the sub-structuring of a single glacial refuge across a wide east-to-west gradient.


Consensus neighbor-joining phylogram depicting D C genetic distance (Cavalli-Sforza and Edwards 1967) among the clusters of eastern hemlock, with Carolina hemlock as an outgroup. The values represent the percent bootstrap support for the nodes over 1,000 replicates
The exact location of eastern hemlock refuges during the last glacial maximum is unclear, although it is plausible that restricted populations of hemlock occurred within a narrow latitudinal band of mixed conifer-northern hardwood forest that existed between northern boreal forests and more southern temperate forests (Delcourt and Delcourt 1987). Hemlock pollen was discovered at a site in the lower Mississippi Alluvial Valley in southwestern Tennessee from approximately 20,000 years BP (Delcourt et al. 1980), and northwestern Georgia from approximately 16,000 BP (Watts 1970). Hemlock pollen may also have been present in small amounts in the Coastal Plain of northeastern North Carolina approximately 20,000–25,000 BP, disappearing from the pollen record until about 10,000 years BP (Whitehead 1973). Between 16,000 and 13,000 years ago, hemlock moved north along the Appalachian mountains and colonized portions of the mid-Atlantic seaboard (Delcourt and Delcourt 1987), including the coastal plain of South Carolina (Watts 1980) and the Delmarva Peninsula (Sirkin et al. 1977). Eastern hemlock colonized New England by 10,000 years BP and the eastern Great Lakes area and New York by about 12,000 years BP; it maintained a primary population center in the Central and Southern Appalachians until approximately 6,000 years BP, when the main area of dominance extended into the Northern Appalachians, New England and southeastern Canada. About that time, it arrived at Lake Superior and the Upper Peninsula of Michigan (Delcourt and Delcourt 1987), reaching its recent maximum distributional range and abundance (Davis 1983).
Regional patterns of genetic variation
Patterns of microsatellite genetic variation across the range of eastern hemlock are not unequivocally consistent with the expectation that populations in refugial areas should harbor higher levels of genetic variation than colonized areas (Provan and Bennett 2008). On one hand, all the inferred genetic clusters are present in the southern part of the species range (the putative refugial zone), compared to a single genetic cluster dominating the populations existing in formerly glaciated territory. A similar pattern of genetic structure is common across species of the southeastern United States, most likely the result of the survival and divergence of genomes in separate refuges through repeated glacial and interglacials (Hewitt 2000). As a result, populations at the trailing edge of a species’ range, such as the genetically divergent Bankhead and Hemlock Bluffs populations of eastern hemlock, often harbor a disproportionate share of the genetic resources of a species (Petit et al. 2003).
At the same time, most measures of eastern hemlock genetic variation are, on average, not significantly different between populations in glaciated and unglaciated areas (Fig. 1; Table 5). Many North American studies report lower genetic diversity in northern populations that expanded from refuges south of the ice sheets (Hewitt 2000). These include more northerly populations of mountain hemlock (Tsuga mertensiana [Bong.] Carr.), which suffered a loss of genetic variation due to post-glacial range expansion (Ally et al. 2000), and lodgepole pine (Pinus contorta Doug. ex Loud.), in which allelic richness is related to time since population founding (Cwynar and Macdonald 1987). This is not a universal pattern, however. In whitebark pine (Pinus albicaulis Engelm.), for example, populations from glaciated areas had levels of genetic diversity similar to those from unglaciated areas, perhaps because its seeds are bird-dispersed (Jorgensen and Hamrick 1997). In Europe, the genetic diversity of Alnus glutinosa (L.) Gaertn. populations increased northwards and westwards away from the species’ putative glacial refuge in the Carpathian Mountains, possibly the result of historic processes related to genetic drift and effective population size (Cox et al. 2011).
Also countering expectations is the fact that eastern hemlock appears to have two main centers of genetic variation, one in the refugial Southern Appalachians region and one in a formerly glaciated region encompassing New York and the New England states (Fig. 2a; Table 3). One possible explanation is the existence of a glacial refuge on the currently submerged continental shelf south of New England, which mastodon fossils (Whitmore et al. 1967) and tree pollen (Emery et al. 1967) suggest was unglaciated forestland. Under this model, hemlock from this refugial population would have colonized New England and southeastern Canada to the north, the northern Great Lakes States and Ontario to the west, and the Appalachian chain as far south as North Carolina and Georgia, making secondary contact with hemlocks moving north from more southerly refuges. This scenario seems unlikely, given that the North genetic cluster (which would have had its origin in the continental shelf refuge under this model) and South Central genetic cluster (potentially descended from a refuge in or near the Southern Appalachians) are highly related (Table 4). It is more likely that these two genetic clusters are associated with refuges that had at least a small degree of historic genetic exchange. Additionally, isopoll maps derived from more than 700 fossil-pollen sites show little evidence of high Tsuga pollen abundances in the Northeast until approximately 10,000 BP, while high pollen abundances existed as early as 14,000 years BP in the Southern Appalachians, extending northward over time into New England (Williams et al. 2004).
A potentially more parsimonious explanation for eastern hemlock’s pattern of genetic variation incorporates a well-documented sudden and drastic decline in abundance throughout most of its range about 5,000 years ago (Bennett and Fuller 2002), consistent with a large-scale pathogen or insect outbreak (Davis 1981b; Allison et al. 1986). Fossil evidence indicates that eastern hemlock experienced mass mortality caused by insect defoliation (Anderson et al. 1986), primarily hemlock looper (Lambdina fiscellaria Guen.) in association with eastern spruce budworm (Choristoneura fumiferana Clemens) (Bhiry and Filion 1996). This was followed by a gradual recovery in abundance over 1,000 or more years (Davis 1981a; Foster and Zebryk 1993), potentially as the species developed widespread host resistance (Davis 1981a; Foster 2000). The existence of such widespread host resistance is supported by the fact that the foliar terpenoid chemistry of eastern hemlock and Carolina hemlock is adapted for defense against defoliating insects such as hemlock looper and eastern spruce budworm, while Asian hemlock species have terpenoid profiles suggesting their foliar chemistry is adapted for defense against sucking insects (Lagalante and Montgomery 2003; Lagalante et al. 2007).
The large-scale prehistoric decline associated with this insect outbreak may have resulted in an extreme genetic bottleneck that is unusual for such a widespread species (Petit et al. 2004), as revealed by the microsatellite signature of inbreeding across the entire eastern hemlock range (Table 2) and for nearly all the sampled populations across its range (Fig. 2c). This pattern is consistent with extensive allozyme inbreeding in the southern (Potter et al. 2008) and western (Zabinski 1992) parts of its range. The intensity of the hemlock decline and the residual abundance of the species varied geographically, with trees surviving in rare but widespread populations (Foster 2000). The fossil-pollen record (Williams et al. 2004) suggests eastern hemlock maintained the highest abundance in the northeastern part of its range, while it became rare in the Southern Appalachians. In fact, southern populations are more inbred on average than northern populations (Table 5), contrary to post-glacial migration expectations but consistent with greater population isolation in the south. Perhaps the decline was less severe in the Northeast, allowing eastern hemlock to maintain most of its existing genetic variability in that region, while a precipitous decline in the Southern Appalachians reduced previously high genetic variation to a level on par with the variation present in the Northeast. While we did not detect the signature of a recent genetic bottleneck for eastern hemlock or any of its populations or genetic clusters, we did find evidence of a recent rangewide population expansion that could have followed such an event, in the form of a significant deficiency of heterozygosity compared to expectations under migration-drift equilibrium. Additionally, the probability of recent population expansion was greater in northern than in southern populations (Table 5), and was positively correlated with population latitude (Table 6), possibly suggesting a strong recovery in the north following the prehistoric decline, or a recent continuation of range expansion in the north, or a combination of both.
Widespread inbreeding across the range of eastern hemlock is also consistent with a pattern of long-distance colonization events and subsequent genetic bottlenecks occurring during post-glacial range expansion (Hewitt 1996; Ibrahim et al. 1996; Bialozyt et al. 2006). While this mode of colonization has been demonstrated at the northwestern limit of eastern hemlock in Wisconsin, where the species has expanded its range relatively recently (Parshall 2002), this process does not explain the relatively higher levels of eastern hemlock inbreeding in the areas nearest its putative glacial refuges. Finally, the self-compatibility of eastern hemlock may influence the inbreeding results, although the degree to which this might be the case is unclear. A controlled-cross study of eastern hemlock did not verify the parentage of putative selfed seedlings using molecular markers, nor did it assess subsequent growth and survival; additionally, the number of seedlings germinated per selfed cone (1.12) was much smaller than per outcrossed cone (6.26) (Bentz et al. 2002). Further work is needed to assess the degree to which selfing might affect inbreeding in natural stands of eastern hemlock.
Genetic composition of isolated populations
The distribution of eastern hemlock includes several peripheral disjunct populations along its southern and western edges; seven of these outliers were sampled for this study. These populations are of particular conservation concern because within-population genetic diversity generally declines and among-population genetic differentiation generally increases from the center of a species’ geographic range to its periphery (Eckert et al. 2008). Loss of genetic diversity in small and isolated populations of tree species is often associated with genetic drift and inbreeding (Jaramillo-Correa et al. 2009), and is predicted to reduce overall population fitness (Reed and Frankham 2003) and the capacity of populations to adapt to environmental change (Willi et al. 2006). Differential adaptive pressures, genetic drift, and mutation, meanwhile, could push reproductively isolated populations toward greater genetic differentiation, leading to the potential for speciation (Slatkin 1987).
Isolated disjunct populations of eastern hemlock appear to encompass significantly less genetic variation than range-interior populations by most measures (Table 5). This is contrary to a study of chloroplast DNA restriction fragment length polymorphisms (RFLPs) that included several eastern hemlock outliers, which did not find any difference in heterogeneity between pooled main-range and unglaciated outlier populations, but included only seven populations from the former category and three from the latter (Wang et al. 1997). At the same time, we found that disjunct populations contained more unique alleles, on average, than main-range populations (Table 5). Particularly noteworthy in the current study is the fact that three highly isolated western populations contained unique alleles, Shades State Park in Indiana with three and Hemlock Cliffs in Indiana and Bankhead National Forest in Alabama with one each (Table 3; Fig. 2b). This seems to suggest the effects of long-term isolation and genetic divergence in this region. These results are consistent with the chloroplast RFLP study, which detected relatively high fragment diversity in isolated populations (Wang et al. 1997).
As expected, disjunct populations of eastern hemlock are more genetically distinct than main-range populations, as measured by mean pairwise genetic distance from a given population to all other populations (Table 5). Additionally, trees from the Bankhead National Forest were genetically distinct enough to have been assigned to a unique genetic cluster (Fig. 3). While the populations in Indiana are glacial relicts where local conditions are not the best suited for eastern hemlock (Friesner and Polzger 1931b), seedlings have been able to establish on steep slopes and other areas with shallow forest litter (Friesner and Polzger 1931a). Eastern hemlocks in the Bankhead disjunct area in Alabama contain viable populations that are successfully regenerating, if highly localized (Hart and Shankman 2005), while eastern hemlock population at Hemlock Bluffs in North Carolina seems to barely be able to survive and reproduce under conditions that approach the xeric extreme of its adaptability (Oosting and Hess 1956). It is therefore not surprising that Hemlock Bluffs is the least genetically diverse population by several measures (Table 3).
Gene conservation implications
Studies of the genetic diversity of forest trees over substantial parts of their distributions are relevant for forest and landscape management, the inventory of genetic resources, and the conservation of rare, endemic, relictual and endangered tree species (Pautasso 2009). In addition to clarifying recent evolutionary history for an ecologically important eastern North American conifer, the results of this range-wide molecular marker study of eastern hemlock are helpful for the development of ongoing ex situ conservation efforts by Camcore (international tree breeding and conservation program at North Carolina State University) and USDA Forest Service’s Forest Health Protection program that have already secured seed from 418 mother trees in 60 populations of eastern hemlock (Camcore 2010). The primary goal of the ex situ conservation effort is the capture of a seed sample that is broadly adapted and that, in response to devastation by hemlock woolly adelgid, can be used to repopulate eastern hemlock populations across the wide variety of habitat types and climate zones found within the geographic range of the species. The molecular marker data presented in this paper will be used to refine the seed collections within putative adaptive zones, determined based on environmental characteristics across the range of the species, so that these collections are representative of the patterns of genetic diversity present in the landscape and are focused on areas with high levels of genetic variation or that contain rare and unique alleles. While selectively neutral markers such as microsatellites may have only limited ability to predict quantitative trait diversity (Reed and Frankham 2001), some evidence suggests that heterozygosity may be associated with fitness (Reed and Frankham 2003) and that neutral marker differentiation among populations could be at least roughly predictive of genetic differentiation among populations in quantitative traits (Leinonen et al. 2008). Although not ideal, molecular genetic diversity is the most rapidly and easily assessable measure of diversity in natural populations, and so remains our best estimate of adaptive potential of these populations in an uncertain environment (Jump et al. 2009).
Gene conservation efforts in the Southern Appalachians and New England are likely to capture high levels of genetic variation in eastern hemlock, including allelic richness and heterozygosity. Ex situ and in situ conservation activities should not be limited to these two regions, however. While some unique alleles are present in these regions, populations with the most unique alleles are located in the western reaches of the eastern hemlock range. This may result in part from a low sampling intensity in the Great Lakes states and elsewhere along the western periphery of the range, but these alleles are still specific to the western region and would not be captured if seed collections were limited to other regions.
Additionally, gene conservation for eastern hemlock should incorporate thorough representation from peripheral populations. While these are less genetically diverse than interior populations for several measures of genetic variation, they also contain more unique alleles on average (Table 5), in keeping with the expectation that conservation of peripheral disjunct populations may present the best opportunity for conserving rare alleles (Gapare et al. 2005). Adequately conserving genetic diversity in peripheral populations, however, may require collections over larger areas than in core populations because of fine-scale genetic structure associated with the lower density of adult trees (Gapare and Aitken 2005). Isolated disjunct eastern hemlock populations also are more genetically distant on average from all other populations (pairwise D C ) than are interior populations (Table 5), and their genetic composition consists largely of genetic clusters other than the widespread North genetic cluster (Fig. 3). In the face of changing climate conditions, these populations may be among the most at risk. While low genetic variation in peripheral populations may not necessarily impair their response to climate change (Pautasso et al. 2010), strong and continuous directional selection under extreme environmental conditions might further reduce their genetic variability for fitness-related traits compared to central populations, which are expected to experience a much larger component of stabilizing selection (Eckert et al. 2008). Also, peripheral populations of eastern hemlock generally are small, some containing fewer than 1,000 trees, while all are isolated from the generally continuous eastern hemlock distribution where considerable short- to middle-distance gene exchange is likely to be common. Individuals in such small and isolated populations are generally less fit as a result of environmental stress and inbreeding, forces that can substantially increase the probability of population extirpation under changing environmental conditions (Willi et al. 2006).
Finally, a comprehensive genetic conservation strategy aiming to adequately sample eastern hemlock genetic variation will need to encompass regions both infested and as yet uninfested by the hemlock woolly adelgid, even though, on average, few significant differences exist in population-level measures of genetic diversity between populations within and outside of counties currently infested by the insect (Table 5). Gene conservation efforts in currently uninfested areas, including the Midwest, western Pennsylvania and New York, northern New York and New England, and Canada (United States Department of Agriculture Forest Service 2011), would capture much allelic diversity. Such efforts are warranted, given that predicted warmer winters could allow HWA to spread into portions of the hemlock range where it is currently unable to survive (Dukes et al. 2009). Infested areas, meanwhile, may contain many unique alleles and novel gene combinations. In fact, the existence of all inferred genetic clusters within the southern part of the eastern hemlock range, compared to only one across much of the north, argues in favor of intense seed collection and stand-management activities in the south, which was almost entirely infested by 2010 (United States Department of Agriculture Forest Service 2011). The need for such gene conservation activities is urgent, given that HWA can kill untreated hemlocks within 4 years (McClure 1991) and that it has spread particularly quickly in recent years through the southern portion of the eastern hemlock distribution (Onken and Reardon 2010).
References
Albani M, Moorcroft PR, Ellison AM et al (2010) Predicting the impact of hemlock woolly adelgid on carbon dynamics of eastern United States forests. Can J For Res 40:119–133
Allen MC, Sheehan J, Master TL et al (2009) Responses to Acadian flycatchers (Empidonax virescens) to hemlock woolly adelgid (Adelges tsugae) infestation in Appalachian riparian forests. Auk 126:543–553
Allison TD, Moeller RE, Davis MB (1986) Pollen in laminated sediments provides evidence for a Mid-Holocene forest pathogen outbreak. Ecology 67:1101–1105
Ally D, El-Kassaby Y, Ritland K (2000) Genetic diversity, differentiation and mating system in mountain hemlock (Tsuga mertensiana) across British Columbia. For Genet 7:97–108
Amarasinghe V, Brown GR, Mank JE et al (2002) Microsatellite DNA loci for westem hemlock [Tsuga heterophylla (Raf.) sarg]. Mol Ecol Notes 2:236–238
Anderson RS, Davis RB, Miller NG et al (1986) History of late-glacial and post-glacial vegetation and disturbance around Upper South Branch Pond, northern Maine. Can J Bot Rev Can Bot 64:1977–1986
Barton NH, Slatkin M (1986) A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56:409–415
Bennett KD, Fuller JL (2002) Determining the age of the mid-Holocene Tsuga canadensis (hemlock) decline, eastern North America. Holocene 12:421–429
Bennett KD, Provan J (2008) What do we mean by refugia? Quat Sci Rev 27:2449–2455
Bentz SE, Riedel LGH, Pooler MR et al (2002) Hybridization and self-compatibility in controlled pollinations of eastern North American and Asian hemlock (Tsuga) species. J Arboric 28:200–205
Besag J (1975) Statistical analysis of non-lattice data. Statistician 24:179–195
Bhiry N, Filion L (1996) Mid-Holocene hemlock decline in eastern North America linked with phytophagous insect activity. Quat Res 45:312–320
Bialozyt R, Ziegenhagen B, Petit RJ (2006) Contrasting effects of long distance seed dispersal on genetic diversity during range expansion. J Evol Biol 19:12–20
Bonner FT, Karrfalt RP (eds) (2008) The woody plant seed manual. U.S. Department of Agriculture, Forest Service, Washington, DC
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455
Brooks RT (2001) Effects of the removal of overstory hemlock from hemlock-dominated forests on eastern redback salamanders. For Ecol Manag 149:197–204
Brown AHD, Hardner CM (2000) Sampling the gene pools of forest trees for ex situ conservation. In: Young AG, Boshier D, Boyle TJ (eds) Forest conservation genetics: principles and practice. CABI Publishing, Collingwood, pp 185–196
Camcore (2010) 2010 Camcore Annual Report. Camcore International Tree Conservation and Domestication, Department of Forestry and Environmental Resources, North Carolina State University, Raleigh, North Carolina, http://www.camcore.org/publications/documents/AnnReport 2010 Online.pdf. Accessed 7 June 2011
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis: models and estimation procedures. Evolution 21:550–570
Cheah C, Montgomery ME, Salom S et al (2004) Biological Control of Hemlock Woolly Adelgid. United States Department of Agriculture, Forest Service, Forest Health Technology Enterprise Team, Morgantown, p 22
Chen C, Durand E, Forbes F et al (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Cobb RC (2010) Species shift drives decomposition rates following invasion by hemlock woolly adelgid. Oikos 119:1291–1298
Cobb RC, Orwig DA, Currie S (2006) Decomposition of green foliage in eastern hemlock forests of southern New England impacted by hemlock woolly adelgid infestations. Can J For Res 36:1331–1341
Comes HP, Kadereit JW (1998) The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci 3:432–438
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cox K, Broeck AV, Van Calster H et al (2011) Temperature-related natural selection in a wind-pollinated tree across regional and continental scales. Mol Ecol 20:2724–2738
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Cwynar LC, Macdonald GM (1987) Geographical variation of lodgepole pine in relation to population history. Am Nat 129:463–469
Davis MB (1981a) Outbreaks of forest pathogens in Quaternary history. In: Birks HJB (ed) Proceedings of the fourth international Palynological conference, 1976–1977. Birbal Sahni Institute of Paleobotany, Lucknow, pp 216–227
Davis MB (1981b) Quaternary history and the stability of forest communities. In: Shugart HH, Botkin DB, West DC (eds) Forest succession, concepts and applications. Springer, New York, pp 132–154
Davis MB (1983) Holocene vegetational history of the Eastern United States. In: Wright HE Jr (ed) Late Quaternary environments of the United States: the holocene. University of Minnesota Press, Minneapolis, pp 166–181
Delcourt PA, Delcourt HR (1987) Long-term forest dynamics of the temperate zone. Springer, New York
Delcourt PA, Delcourt HR, Brister RC et al (1980) Quaternary vegetation history of the Mississippi embayment. Quat Res 13:111–132
Dukes JS, Pontius J, Orwig D et al (2009) Responses of insect pests, pathogens, and invasive plant species to climate change in the forests of northeastern North America: what can we predict? Can J For Res 39:231–248
Durand E, Jay F, Gaggiotti OE et al (2009) Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26:1963–1973
Dyke AS, Moore A, Robertson L (2003) Deglaciation of North America, geological survey of Canada, Open File 1574, Ottawa, Canada, Map, presentation scale 1:30,000,000, colored. [Also on CD-ROM as a GIS coverage in ArcINFO format]
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Eickmeier W, Adams M, Lester D (1975) Two physiological races of Tsuga canadensis in Wisconsin. Can J Bot 53:940–951
Ellison AM, Bank MS, Clinton BD et al (2005) Loss of foundation species: consequences for the structure and dynamics of forested ecosystems. Front Ecol Environ 3:479–486
Emery KO, Wigley RL, Bartlett AS et al (1967) Freshwater peat on the continental shelf. Science 158:1301–1307
Eriksson G, Namkoong G, Roberds JH (1993) Dynamic gene conservation for uncertain futures. For Ecol Manag 62:15–37
ESRI (2006) ArcMap 9.2. Environmental Systems Research Institute Inc., Redlands
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinformatics Online 1:47–50
Farjon A (1990) Pinaceae: drawings and descriptions of the genera Abies, Cedrus, Pseudolarix, Keteleeria, Nothotsuga, Tsuga, Cathaya, Pseudostuga, Larix and Picea. Koeltz Scientific Books, Konigstein
Felsenstein J (2005) PHYLIP (Phylogeny Inference Package), version 3.6. Department of Genome Sciences, University of Washington
Ford CR, Vose JM (2007) Tsuga canadensis (L.) Carr. mortality will impact hydrologic processes in southern appalachian forest ecosystems. Ecol Appl 17:1156–1167
Foster DR (2000) Hemlock’s future in the context of its history: an ecological perspective. In: McManus KA, Shields KS, Souto DR (eds) Proceedings: symposium on sustainable management of Hemlock Ecosystems in Eastern North America. United States Department of Agriculture, Forest Service, Durham, pp 1–4
Foster DR, Zebryk TM (1993) Long-term vegetation dynamics and disturbance history of a Tsuga-dominated forest in New England. Ecology 74:982–998
Francois O, Durand E (2010) Spatially explicit Bayesian clustering models in population genetics. Mol Ecol Resour 10:773–784
Friesner RC, Polzger JE (1931a) Studies in forest ecology I: factors concerned in hemlock reproduction in Indiana. Butler University Botanical Studies 2:133–144
Friesner RC, Polzger JE (1931b) Studies in forest ecology II: the ecological significance of Tsuga canadensis in Indiana. Butler Univ Bot Stud 2:145–149
Gapare WJ, Aitken SN (2005) Strong spatial genetic structure in peripheral but not core populations of Sitka spruce [Picea sitchensis (Bong.) Carr.]. Mol Ecol 14:2659–2667
Gapare WJ, Aitken SN, Ritland CE (2005) Genetic diversity of core and peripheral Sitka spruce (Picea sitchensis (Bong.) Carr) populations: implications for conservation of widespread species. Biol Conserv 123:113–123
Godman RM, Lancaster K (1990) Eastern Hemlock. In: Burns RM, Honkala BH (eds) Silvics of North America: 1 conifers. United States Department of Agriculture Forest Service, Washington DC
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467
Hart JL, Shankman D (2005) Disjunct eastern hemlock (Tsuga canadensis) stands at its southern range boundary. J Torrey Bot Soc 132:602–612
Heard MJ, Valente MJ (2009) Fossil pollen records forecast response of forests to hemlock woolly adelgid invasion. Ecography 32:881–887
Heuertz M, Fineschi S, Anzidei M et al (2004) Chloroplast DNA variation and postglacial recolonization of common ash (Fraxinus excelsior L.) in Europe. Mol Ecol 13:3437–3452
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Holmes JA (1883) Occurrence of Abies canadensis and Pinus strobus in central North Carolina. J Elisha Mitch Sci Soc 1:86–87
Hubisz MJ, Falush D, Stephens M et al (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332
Humphrey LD (1989) Life history traits of Tsuga caroliniana Engelm. (Carolina hemlock) and its role in community dynamics. Castanea 54:172–190
Ibrahim KM, Nichols RA, Hewitt GM (1996) Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77:282–291
Jackson ST (1991) Pollen representation of vegetational patterns along an elevational gradient. J Veg Sci 2:613–624
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jaramillo-Correa JP, Beaulieu J, Khasa DP et al (2009) Inferring the past from the present phylogeographic structure of North American forest trees: seeing the forest for the genes. Can J For Res 39:286–307
Jenkins JC, Aber JD, Canham CD (1999) Hemlock woolly adelgid impacts on community structure and N cycling rates in eastern hemlock forests. Can J Forest Res 29:630–645
Jetton RM, Dvorak WS, Whittier WA (2008a) Ecological and genetic factors that define the natural distribution of Carolina hemlock in the southeastern United States and their role in ex situ conservation. For Ecol Manag 255:3212–3221
Jetton RM, Whittier WA, Dvorak WS et al (2008b) Status of ex situ conservation efforts for Carolina and eastern hemlock in the southeastern United States. In: Onken B, Reardon R (eds) Proceedings of the fourth symposium on Hemlock Woolly adelgid in the Eastern United States. USDA Forest Service, Hartford, pp 81–89
Jetton RM, Hastings FL, Hain FP (2009) Arthropod diversity associated with old and secondary growth eastern hemlock (Tsuga canadensis) in the Great Smoky Mountains National Park. J N C Acad Sci 125:70–77
Jorgensen SM, Hamrick JL (1997) Biogeography and population genetics of whitebark pine, Pinus albicaulis. Can J For Res 27:1574–1585
Josserand SA, Potter KM, Echt CS et al (2008) Isolation and characterization of microsatellite markers for Carolina hemlock (Tsuga caroliniana). Mol Ecol Resour 8:1371–1374
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jump AS, Marchant R, Penuelas J (2009) Environmental change and the option value of genetic diversity. Trends Plant Sci 14:51–58
Karhu A, Vogl C, Moran GF et al (2006) Analysis of microsatellite variation in Pinus radiata reveals effects of genetic drift but no recent bottlenecks. J Evol Biol 19:167–175
Kessell SR (1979) Adaptation and dimorphism in eastern hemlock, Tsuga canadensis (L.) Carr. Am Nat 113:333–350
Kimura M, Crow JF (1964) Number of alleles that can be maintained in a finite population. Genetics 49:725–738
Kimura M, Ohta T (1978) Stepwise mutation model and distribution of allelic frequencies in a finite population. Proc Natl Acad Sci USA 75:2868–2872
Lagalante AF, Montgomery ME (2003) Analysis of terpenoids from hemlock (Tsuga) species by solid-phase microextraction/gas chromatography/ion-trap mass spectrometry. J Agric Food Chem 51:2115–2120
Lagalante AF, Montgomery ME, Calvosa FC et al (2007) Characterization of terpenoid volatiles from cultivars of eastern hemlock (Tsuga canadensis). J Agric Food Chem 55:10850–10856
Leinonen T, O’Hara RB, Cano JM et al (2008) Comparative studies of quantitative trait and neutral marker divergence: a meta-analysis. J Evol Biol 21:1–17
Lemieux MJ, Beaulieu J, Bousquet J (2011) Chloroplast DNA polymorphisms in eastern hemlock: range-wide genogeographic analyses and implications for gene conservation. Can J For Res 41:1047–1059
Libiger O, Nievergelt CM, Schork NJ (2009) Comparison of genetic distance measures using human SNP genotype data. Hum Biol 81:389–406
Little EL (1971) Atlas of United States trees, vol 1. Conifers and Important Hardwoods, Washington, DC
Luikart G, Allendorf FW, Cornuet JM et al (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
McClure MS (1991) Density-dependent feedback and population cycles in Adelges tsugae (Homoptera, Adelgidae) on Tsuga canadensis. Environ Entomol 20:258–264
McClure MS, Salom SM, Shields KS (2003) Hemlock Woolly Adelgid. Forest Health Technology Enterprise Team, United States Department of Agriculture, Forest Service, Morgantown, p 14
McLachlan JS, Clark JS, Manos PS (2005) Molecular indicators of tree migration capacity under rapid climate change. Ecology 86:2088–2098
Mihaescu R, Levy D, Pachter L (2009) Why neighbor-joining works. Algorithmica 54:1–24
Montgomery ME, Bentz SE, Olsen RT (2009) Evaluation of hemlock (Tsuga) species and hybrids for resistance to Adelges tsugae (Hemiptera: Adelgidae) using artificial infestation. J Econ Entomol 102:1247–1254
Nienstaedt H, Kriebel HB (1955) Controlled pollination of eastern hemlock. For Sci 1:115–120
Nienstaedt H, Olson JS (1961) Effects of photoperiod and source on seedling growth of eastern hemlock. For Sci 7:81–96
Nuckolls AE, Wurzburger N, Ford CR et al (2009) Hemlock declines rapidly with hemlock woolly adelgid infestation: impacts on the carbon cycle of southern Appalachian forests. Ecosystems 12:179–190
O’Connell LM, Ritland K, Thompson SL (2008) Patterns of post-glacial colonization by western redcedar (Thuja plicata, Cupressaceae) as revealed by microsatellite markers. Botany 86:194–203
Olson JS, Nienstaedt H (1957) Photoperiod and chilling control growth of hemlock. Science 125:492–494
Onken B, Reardon R (eds) (2010) Proceedings of the fifth symposium on Hemlock Woolly adelgid in the Eastern United States. USDA Forest Service, Forest Health Technology Enterprise Team, Morgantown
Oosting HJ, Hess DW (1956) Microclimate and a relic stand of Tsuga canadensis in the lower piedmont of North Carolina. Ecology 37:28–39
Orwig DA, Foster DR (1998) Forest response to the introduced hemlock woolly adelgid in southern New England, USA. J Torrey Bot Soc 125:60–73
Parshall T (2002) Late holocene stand-scale invasion by hemlock (Tsuga canadensis) at its western range limit. Ecology 83:1386–1398
Pautasso M (2009) Geographical genetics and the conservation of forest trees. Perspect Plant Ecol Evol Syst 11:157–189
Pautasso M, Dehnen-Schmutz K, Holdenrieder O et al (2010) Plant health and global change—some implications for landscape management. Biol Rev 85:729–755
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petit RJ, Brewer S, Bordacs S et al (2002) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manag 156:49–74
Petit RJ, Aguinagalde I, de Beaulieu JL et al (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Petit RJ, Bialozyt R, Garnier-Gere P et al (2004) Ecology and genetics of tree invasions: from recent introductions to Quaternary migrations. For Ecol Manag 197:117–137
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Potter KM, Dvorak WS, Crane BS et al (2008) Allozyme variation and recent evolutionary history of eastern hemlock (Tsuga canadensis) in the southeastern United States. New For 35:131–145
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Provan J, Bennett KD (2008) Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol 23:564–571
Raymond M, Rousset F (1995) Genepop (Version-1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reed DH, Frankham R (2001) How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evolution 55:1095–1103
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Rohr JR, Mahan CG, Kim KC (2009) Response of arthropod biodiversity to foundation species declines: the case of the eastern hemlock. For Ecol Manag 258:1503–1510
Ross RM, Bennett RM, Snyder CD et al (2003) Influence of eastern hemlock (Tsuga canadensis L.) on fish community structure and function in headwater streams of the Delaware River basin. Ecol Freshw Fish 12:60–65
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
SAS Institute Inc. (2008) The SAS system for Windows, Version 9.2. Cary
Schaberg PG, DeHayes DH, Hawley GJ et al (2008) Anthropogenic alterations of genetic diversity within tree populations: implications for forest ecosystem resilience. For Ecol Manag 256:855–862
Shamblin BM, Faircloth BC, Josserand SA et al (2008) Microsatellite markers for eastern hemlock (Tsuga canadensis). Mol Ecol Resour 8:1354–1356
Siderhurst LA, Griscom HP, Hudy M et al (2010) Changes in light levels and stream temperatures with loss of eastern hemlock (Tsuga canadensis) at a southern Appalachian stream: implications for brook trout. For Ecol Manag 260:1677–1688
Sirkin LA, Denny CS, Rubin M (1977) Late Pleistocene environment of central Delmarva Peninsula, Delaware-Maryland. Geol Soc Am Bull 88:139–142
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Snyder CD, Young JA, Lemarie DP et al (2002) Influence of eastern hemlock (Tsuga canadensis) forests on aquatic invertebrate assemblages in headwater streams. Can J Fish Aquat Sci 59:262–275
Spaulding HL, Rieske LK (2010) The aftermath of an invasion: structure and composition of Central Appalachian hemlock forests following establishment of the hemlock woolly adelgid, Adelges tsugae. Biol Invasions 12:3135–3143
Taberlet P, Fumagalli L, Wust-Saucy AG et al (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Takezaki N, Nei M (1996) Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144:389–399
Templer PH, McCann TM (2010) Effects of the hemlock woolly adelgid on nitrogen losses from urban and rural northern forest ecosystems. Ecosystems 13:1215–1226
Tingley MW, Orwig DA, Field R (2002) Avian response to removal of a forest dominant: consequences of hemlock woolly adelgid infestations. J Biogeogr 29:1505–1516
United States Department of Agriculture Forest Service (2011) Counties with established HWA populations, 2010. United States Department of Agriculture Forest Service, Forest Health Protection, http://na.fs.fed.us/fhp/hwa/maps/2010.pdf. Accessed 8 Feb 2011
van Oosterhout C, Hutchinson WF, Wills DPM et al (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wang C, Perlin MH, VanStockum RR et al (1997) Chloroplast DNA polymorphisms in Tsuga canadensis and Tsuga caroliniana. Can J For Res 27:1329–1335
Ward JS, Montgomery ME, Cheah C et al (2004) Eastern Hemlock forests: guidelines to minimize the impacts of Hemlock Woolly Adelgid. United States Department of Agriculture, Forest Service, Morgantown, p 27
Watts WA (1970) Full-glacial vegetation of northwestern Georgia. Ecology 51:17–33
Watts WA (1980) Late-Quaternary vegetation history at White Pond on the inner coastal plain of South Carolina. Quat Res 13:187–199
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wellman H, Pritchard E, Benowicz A et al (2003) Microsatellite markers in western hemlock [Tsuga heterophylla (Raf.) Sarg]. Mol Ecol Notes 3:592–594
Whitehead DR (1973) Late-Wisconsin vegetational changes in unglaciated eastern North America. Quat Res 3:621–631
Whitmore FC, Emery KO, Cooke HBS et al (1967) Elephant teeth from Atlantic continental shelf. Science 156:1477–1481
Willi Y, Van Buskirk J, Hoffmann AA (2006) Limits to the adaptive potential of small populations. Annu Rev Ecol Evol Syst 37:433–458
Williams JW, Shuman BN, Webb T et al (2004) Late-Quaternary vegetation dynamics in North America: scaling from taxa to biomes. Ecol Monogr 74:309–334
Zabinski C (1992) Isozyme variation in eastern hemlock. Can J For Res 22:1838–1842
Acknowledgments
We thank Sedley Josserand for her advice in optimizing microsatellite PCRs; Joe Nairn and Brian Shamblin for providing eastern hemlock microsatellite primer sequences; John Frampton and Dana Nelson for assistance in initiating this project; Ricardo Hernandez and Jennifer DeWoody for laboratory assistance; and Craig Echt, Alistair Jump and two anonymous reviewers for thoughtful comments on the manuscript. We are very grateful to a long list of government organizations listed in Table 1 and their natural resource professionals who assisted with the identification and collection of hemlock samples. This work was supported by in part through Grant Agreements 05-DG-11083150-210 and 08-DG-11083150-014 and Participating Agreement 06-PA-11083150-002 between the U.S. Department of Agriculture, Forest Service, Forest Health Protection and Camcore, North Carolina State University; and through Cooperative Agreement 05-CA-11330126-210 and Research Joint Venture Agreements 08-JV-11330146-078 and 10-JV-11330146-049 between the U.S. Department of Agriculture, Forest Service, Southern Research Station, and North Carolina State University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
10592_2011_301_MOESM1_ESM.xls
Online Resource 1: Population pairwise chord genetic distance (D C ) for 60 eastern hemlock populations, based on 13 polymorphic nuclear microsatellites (XLS 94 kb)
10592_2011_301_MOESM2_ESM.xls
Online Resource 2: Population pairwise differentiation values (F ST ) for 60 eastern hemlock populations, based on 13 polymorphic nuclear microsatellites (XLS 96 kb)
Rights and permissions
About this article
Cite this article
Potter, K.M., Jetton, R.M., Dvorak, W.S. et al. Widespread inbreeding and unexpected geographic patterns of genetic variation in eastern hemlock (Tsuga canadensis), an imperiled North American conifer. Conserv Genet 13, 475–498 (2012). https://doi.org/10.1007/s10592-011-0301-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0301-2