
Abstract
Montane regions can promote allopatric speciation and harbor unique species with small ranges. The southern Appalachians are a biodiversity hotspot for salamanders, and several montane endemics occur in the region. Here, we present the first DNA sequence data for Plethodon sherando, a terrestrial salamander recently discovered in the Blue Ridge Mountains of Virginia. We sequenced two mitochondrial regions (cyt-b and CO1) from salamanders at reference sites near the center of P. sherando’s range and from two contact zones where P. sherando populations are replaced by Plethodon cinereus, the Northern Red-Backed salamander. We then used these sequence data to examine divergence and hybridization between the two taxa. We found P. sherando and P. cinereus morphotypes from contact zones to be reciprocally monophyletic and highly divergent (~17%). P. sherando exhibited very low sequence diversity (π = 0.0010) as compared to P. cinereus from the same locations (π = 0.0096). Salamander morphology in the contact zone was as distinct as morphology at reference sites, and discriminant function analysis based on morphology successfully classified 98% of salamanders to their mitochondrial lineage. Phylogenetic analysis of cyt-b sequences showed P. sherando to be sister to Plethodon serratus (the Southern Red-Backed salamander) rather than P. cinereus or any nearby mountaintop endemics. Our results suggest that P. sherando is a distinct lineage that is not subject to substantial introgression from P. cinereus and that may have a history of geographic isolation. Given its limited range (<80 km2), we believe P. sherando should merit a conservation status similar to that of other mountaintop salamanders in the region.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Montane regions are important biodiversity hot spots for a variety of plant and animal groups (Anderson and Maldonado-Ocampo 2010; McCain 2005; Myers et al. 2000). Mountain ecosystems can also be important factors in the creation of biodiversity, with mountain ranges providing opportunities for allopatric speciation (Knowles 2001; Steele et al. 2005; Xu et al. 2010). This may be particularly true in the tropics, where changes in elevation have been shown to correlate with different species assemblages or phenotypic traits (e.g. Lovett 1998; Terborgh 1977; Kleindorfer et al. 2006). However, temperate montane regions can also provide opportunities for speciation, and many species are endemic to montane regions of the temperate zone (Kozak and Wiens 2006).
Montane endemics may present particular challenges for conservation. For one, these species may be very susceptible to climate change since they cannot easily shift their ranges northwards or upslope in response to climate warming (Lawler et al. 2009; Moritz et al. 2008; Wilson et al. 2007). Studies that model shifts in habitat distribution under climate change projections generally project range contractions or extinctions for montane species (LaSorte and Jetz 2010; Milanovich et al. 2010). And for many montane species, replacement by competing species with higher thermal tolerances is potentially an even greater threat than simple change in the distribution of suitable habitats (Gilman et al. 2010).
In addition, mountaintop endemics, like other spatially-restricted species, may be susceptible to genetic swamping if they hybridize more widespread species. Gene flow at the boundaries of two related species is relatively common and does not necessarily threaten the coexistence of the two species (Colliard et al. 2010; Mallet 2005; Mallet et al. 2007). However, in the case of rare or spatially restricted species, introgression can progress to the point where the gene pool of the rare species is essentially subsumed into the gene pool of the more common species (Chan et al. 2006; Hegde et al. 2006; Rhymer and Simberloff 1996). Thus, the magnitude of gene flow between montane endemics and their congeners can influence whether or not species-specific adaptive genetic complexes will be maintained over longer time periods.
The southern Appalachians are considered a biodiversity hotspot for terrestrial salamanders of the family Plethodontidae (Duellman and Sweet 1999; Milanovich et al. 2010; Rissler and Smith 2010). The largest Plethodontid genus, Plethodon, is divided into two subgroups—a western group of nine species and an eastern group of at least 46 distinct species, some of them cryptic (Wiens et al. 2006). These salamanders have limited dispersal ability (Cabe et al. 2007; Marsh et al. 2008) and may exhibit niche conservatism over evolutionary time (Kozak and Wiens 2010), factors which have likely contributed to the evolution of mountaintop species with very small ranges. For example, the Peaks of Otter salamander (Plethodon hubrichti), is restricted to forest habitats above 845 m within a narrow 19 km long stretch of the Blue Ridge Mountains in Virginia (Petranka 1998). The Cheat Mountain salamander, Plethodon nettingi, is a federally-threatened species found in a number of small populations on the high Cheat Mountain ridge in West Virginia (Pauley 2007). The Shenandoah salamander, Plethodon shenandoah, which is listed as federally endangered, is found in talus outcrops on three mountains in Shenandoah National Park (Highton and Worthington 1967). These species are notable in having among the smallest ranges of any mainland vertebrates in North America (Carpenter et al. 2001; Highton 1995).
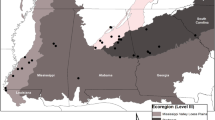
Recently, a new endemic mountaintop Plethodon was identified in the southern Appalachians. Plethodon sherando, the Big Levels salamander, was proposed as a new species on the basis of morphological differences such as longer limbs and fewer trunk vertebrae in comparison to the widespread species Plethodon cinereus (Northern Red-Backed salamander) that surrounds it (Highton 2004). In addition, Highton (2004) reported differences in allozyme frequencies between P. sherando and P. cinereus. Salamanders that are morphologically P. sherando have a range that apparently consists of less than 80 km2 along the top of the Big Levels area of Augusta county, Virginia (Highton 2004). P. sherando is found along ridgetops and on the slopes of Big Levels down to about 640–670 m (Fig. 1), where populations abruptly transition to animals that appear to be P. cinereus (Highton 2004). An additional contact zone is found at high elevations, where Big Levels wraps around to meet the main Blue Ridge (Fig. 1). Little is currently known about P. sherando aside from the general outlines of its range and some morphological characters that distinguish it from P. cinereus. The species is absent from recent analyses of the phylogeny and evolution of the Plethodon genus (e.g. Kozak and Wiens 2006, 2010; Wiens et al. 2006). Moreover, unlike the other mountaintop endemics in the region, P. sherando has no special conservation designation, primarily because of its recent discovery and uncertainty about its status.

Map of the range of P. sherando and our sampling sites. Filled circles indicate sites where P. sherando was found by Highton (2004) or in our own surveys. “X”s indicate sites where P. cinereus but not P. sherando was found. PS1 and PS2 indicate P. sherando reference sites one and two, PC1 and PC2 indicate P. cinereus reference sites, and CZ1 and CZ2 are two contact zone sites where both species were found
We used sequence data from two mitochondrial regions and morphological data across two contact zones to examine genetic diversity and differentiation in P. sherando and the relationship between this salamander and the widespread species P. cinereus that surrounds it. More specifically, we sought to determine: (1) whether animals that are morphologically P. sherando versus P. cinereus are reciprocally monophyletic, (2) if morphological intermediates between P. sherando and P. cinereus are found in contact zones at low or high elevation, (3) if morphology and mitochondrial genotype are concordant across individuals in contact zones, and (4) the phylogenetic relationship between P. sherando and other salamanders in the P. cinereus group.
Methods
The range of P. sherando is centered on the Big Levels ridges of Augusta County, VA (type locality W37°55′09″, N79°04′00″). Along these ridges, P. sherando has been found down to an elevation of about 600 m, though most contact zones with P. cinereus appear closer to 660 m. Based on Highton’s (2004) collection locations as well as our own surveys, the likely range for P. sherando appears to be between 63 km2 and 73 km2 assuming a lower elevational limit of 600 and 660 m respectively (Fig. 1).
For our study, P. sherando and P. cinereus samples were collected from two transects in the Big Levels region (Fig. 1). Each transect consisted of a P. cinereus reference site, a contact zone containing both species, and a P. sherando reference site. Reference sites were identified based on the surveys reported in Highton (2004), whereas contact zones were identified from our own surveys. The first transect covered a typical elevational gradient for these species, running from a P. cinereus reference site (PC1, Fig. 1) at the base of the ridge (580 m) through a mid-elevation (650–670 m) contact zone (CZ1) to a ridgetop P. sherando reference site (PS1) at 1,020 m. The second transect remained at a similar elevation (950–1,050 m) for all three sites, running from a P. cinereus reference site on the Blue Ridge (PC2), through a contact zone where the Blue Ridge connects to Big Levels (CZ2), and terminating in a P. sherando reference site within Big Levels (PS2, Fig. 1). For comparative purposes, we also collected six samples of P. hubrichti, the Peaks of Otter salamander from a single site within its mountaintop range approximately 40 km to the southwest. We considered this to be a relevant comparison because P. hubrichti has a similar range size to that of P. sherando, and it is likewise surrounded by P. cinereus at lower elevations.
Collection and morphological measurements
We located salamanders by turning over rocks and logs along the forest floor and searching through the leaf litter. Searches were carried out during the day in April–May and September–October, 2009. When we captured salamanders, we placed them in plastic bags and cooled them before morphological measurements were taken on live animals.
We used morphological features identified in Highton’s (2004) original study for distinguishing P. sherando from P. cinereus. P. sherando typically has a wider head, longer limbs, and a shorter trunk than P. cinereus. Because P. sherando has longer legs and a shorter trunk than P. cinereus, the number of intercostal spaces (i.e. the spaces between the ribs, hereafter ICS) when limbs are pressed flat against the body is lower in P. sherando than in P. cinereus. When corrected for snout-vent length (SVL), this metric shows little or no overlap between taxa (Highton 2004) with P. sherando normally having an ICS/SVL ratio <0.13 and P. cinereus having a ratio >0.15. Because measurements were taken on live animals, some measurement error was expected. To minimize this error, each measurement was recorded independently by 2–3 different observers and these measurements were averaged for each animal.
After the morphological measurements for each animal were recorded, we collected a 0.5 cm tail tip with forceps from 12 to 16 animals per species per site. Salamanders were then released at the site where they were collected. Plethodontid salamanders lose tails naturally as an antipredator defense (Jaeger and Forester 1993), and tails tips typically regrow within several weeks. Tissue samples from tail tips were stored in collection buffer (10 mM Tris, 10 mM EDTA, pH 8) at 4°C before DNA extraction (Cabe et al. 2007). Each of the 12–16 samples from each site was sequenced for both CO1 and cyt-b.
DNA sequencing
Genomic DNA extraction was performed using the methods described in Connors and Cabe (2003) and Cabe et al. (2007). Genomic DNA was extracted from tissue samples within 24 h of collection using the Promega Wizard® Genomic DNA Purification Kit; (Madison, WI) and quantified using a NanoDrop® spectrophotometer (Wilmington, DE).
We amplified and sequenced segments of the cytochrome oxidase I (CO1) and cytochrome B (cyt-b) genes. We chose CO1 to provide information for the Barcode of Life project (Smith et al. 2008), and we chose cyt-b because this gene has been used frequently in prior phylogenetic analyses of the Plethodon genus (Wiens et al. 2006).
PCR was used to amplify a 750 bp fragment of the CO1 using LepF1-T3 (a T3 tailed version of the LepF1 primer, AATTAACCCTCACTAAAGATTCAACCAATCATAAAGATATTGG′) and LepR1 (5′-TAAACTTCTG GATGTCCAAAAAATCA-3′) or LepF1 (ATTCAACCAATCATAAAGATATTGG) and LepR1-T7 (TAATACGACTCACTATAGGGTAAACTTCTGGATGTCCAAAAAATCA) following Hebert et al. (2004) and Smith et al. (2008). CO1 PCR reactions (50 μl) contained 1× GoTaq Buffer®, 1.5 mM MgCl2, 200 μM dNTP’s, 0.5 μM LepF1-T3, 0.5 μM Lep-R1, 1.25 U/50 μl Taq Polymerase, and 0.02–0.4 ng/μl genomic DNA as template, using the GoTaq® PCR Core kit (Promega, Madison, WI). Cycling conditions were 94°C for 5 min followed by 34 cycles of 94°C for 30 s, 53.5°C for 1 min, and 72°C for 1.5 min, and a final extension at 72°C for 5 min.
A portion of the cytochrome B gene was amplified using a T3 tailed primer PcCytB-F-T3 (5′-AATTAA CCCTCACTAAAGGGCTCAACCAAAACCTTTGACC-3′) and PcCytB-R (5′-TAGCCCCCAATTTTGGTTTACA-3′), both of which were designed using the complete P. cinereus mtDNA sequence available in GenBank (accession number AY728232.1). Cyt-b reactions mirrored CO1 reactions, except that primers annealed at 47°C for 45 s. Products from successful PCR reactions were purified using the Wizard SV Gel and PCR Clean up System (Promega, Madison, WI). Sequencing reactions for both genes were generated using an IR700 labeled T3 primer and the SequiTherm Excel® II Sequencing Kit-LC (Epicenter, Madison, WI). The PCR products were sequenced using a LiCor 4300 DNA analyzer and base calls automatically scored with eSeq v. 3.1 (LiCor, Lincoln, NE). A subset of CO1 sequences was validated by sequencing with an IR800 labeled T7 primer. Sequences contributing to our analysis are archived in GenBank as samples JF731278–JF31333.
Sequence alignment, distance, and diversity
Sequences for each gene were manually edited, trimmed, and aligned using ClustalW in MEGA 4 (Tamura et al. 2007). Alignments were unambiguous (i.e. no gaps were required) and resulted in a 684 bp segment of cyt-b and a 599 bp segment of CO1. Some sequences did contain ambiguous bases, particularly near the ends of the sequences. Ambiguous bases were treated as missing data in all analyses and unique haplotypes were not classified based on ambiguous bases. In principle, ambiguities can be caused by mixtures of mitochondrial genes and nuclear copies (Bensasson et al. 2001). However, in 26 samples that were sequenced in both directions the same ambiguities were never detected. Furthermore, no stop codons or in-dels were found when individual sequences were translated, suggesting that we successfully amplified mtDNA rather than nuclear copies of mitochondrial sequences. The two segments were concatenated for the analyses below (1,283 bp) unless otherwise specified.
We used the program DnaSP v5 (Rozas et al. 2003) to calculate measures of mtDNA sequence diversity. For the full set of putative P. sherando samples, we calculated haplotype diversity (h, the probability that two randomly chosen haplotypes differ) and nucleotide diversity (π, average number of pairwise differences per nucleotide site). We also calculated these measures for haplotypes sampled exclusively from the two contact zones for both P. sherando and P. cinereus. This allowed us to compare sequence diversity between the two lineages over the same spatial scale.
We compared potential models of nucleotide substitution using jModeltest 0.1 (Posada 2008). For both genes, model selection based on AIC yielded a generalized time reversible model with rate variation among sites with a model weight >0.90 (GTR + Γ, model-averaged Γ = 0.78). We used this model in analyses of genetic parameters, phylogenetic reconstruction, and time to most recent common ancestor (see below).
Phylogenetic reconstruction
We estimated relationships among haplotypes using maximum likelihood (ML) and Bayesian methods, implemented in PHYLIP 3.69 (Felsenstein 1993) and Mr. Bayes 3.1.2 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003), respectively. For computational efficiency, these analyses were carried out on a subset of 24 samples that included at least two samples from each lineage at each site (two contact zone sites plus two reference sites for both P. sherando and P. cinereus).
We first estimated relationships among concatenated cyt-b and CO1 haplotypes. We used genbank sequences for Plethodon petraeus (the Pigeon Mountain salamander; accession numbers AY728222.1, NC_006334.1) as an outgroup. Because CO1 sequences are not available for most Plethodon, we also estimated relationships among cyt-b haplotpyes for an expanded set of species, including P. shenandoah (the Shenandoah salamander; AY378043.1, AY378044.1, AY378045.1, AY378046.1), and Plethodon serratus (the Southern Red-Backed salamander; DQ994981.1, DQ994982.1).
For maximum likelihood analyses, we used four gamma rate categories and a search strategy that included iterations of branch length in all topologies. Five hundred bootstrap replicates were used to test the support for each node. For the Bayesian analyses, we used 1,000,000 MCMC generations with samples taken every 100 time steps. The first 25% of the parameter estimates and trees were discarded as burn-in. Four independent runs from different random starting trees produced nearly identical parameter estimates, and in all four runs the log-likelihood plot had stabilized by 250,000 generations.
Time to most recent common ancestor
Time to the most recent common ancestor (MRCA) for P. sherando and P. cinereus was estimated for unique haplotypes using a coalescent-based approach in BEAST 1.5.4. (Drummond and Rambaut 2007). We used a strict molecular clock model (Tajima’s relative rates test, P = 0.19). We took mean and standard deviation rates of evolution from the fossil-calibrated plethodontid phylogeny of Mueller (2006): 1.04 (±0.27) substitutions per 100 million years for CO1 and 0.62 (±0.16) substitutions per 100 million years for cyt-b. MCMC parameters were set to a chain length of 20 million with a burn-in of 2 million and parameters logged every 1,000 trees. Runs from four different sets of random initial parameter values yielded the same distribution for time to MRCA, suggesting that convergence had been achieved.
Morphological analysis
We conducted several analyses of morphology to examine the distribution of phenotypes across contact zones and reference sites. First, we used cluster analysis to identify the optimal number of clusters based on morphological measurements (interspaces per unit snout-vent length and head width per unit snout-vent length). Clustering was conducted separately for P. sherando and P. cinereus reference sites and for the contact zones between the two species. To assess the validity of grouping salamanders into two species, we compared the fit of a two-cluster model to the fit of a single cluster model or a three cluster model. Fits were compared using Bayesian Information Criteria (BIC) in the R program Mclust 3.0 (Fraley and Raftery 2006).
Following this initial cluster analysis, we used linear discriminant function analysis (LDFA) in SPSS v. 17.0 (SPSS Inc., Chicago, IL) to determine the extent to which salamanders from the P. sherando and P. cinereus reference sites were morphologically distinct from one another. We created a linear discriminant function for location (P. sherando or P. cinereus reference site) based on 2 predictor variables—intercostal spaces per unit snout-vent length, head width per unit SVL. We then used “leave-one-out” cross validation (Hastie et al. 2001) to assess the performance of the discriminant function. In this procedure, each point is left out of the model, then reassigned to a location (P. sherando or P. cinereus site) based on its morphology. These assignments can then be compared to the true origin of the animal in order to assess the ability of the model to discriminate animals from the different classes of sites.
We carried out an analogous LDFA on contact zone samples for which we also had mtDNA sequences. Samples were assigned to species based on the morphological variables given above. These species assignments were then validated by comparing them to the lineage to which their mtDNA had been assigned. If there is little introgression between the lineages and their morphology is distinct, assignment accuracy should be close to 100%. Alternatively, extensive introgression between lineages would be expected to produce some individuals with morphology that is non-concordant with mtDNA genotype.
We additionally carried out a two-way analysis of variance on continuous morphological characters (interspaces/SVL and head width/SVL) for both contact zone and reference site samples. Independent variables were site type (reference or contact zone) and mtDNA lineage (P. sherando or P. cinereus). An interaction between site type and lineage would indicate that morphological differences between mtDNA lineages in contact zones differed in magnitude from those differences at reference sites. If morphological differences were less pronounced at contact zones, this would suggest introgression between lineages.
Population history
We examined the genetic structure of P. cinereus in contact zones versus reference sites and ask whether contact zones might represent areas where P. cinereus have recently infringed on the range of P. sherando. In general, in areas of recent expansion, one would expect lower genetic diversity, an excess of rare haplotypes given the level of nucleotide diversity (i.e., negative values for Fu’s Fs), and a unimodal mismatch distribution (Harpending 1994; Johnson et al. 2007; Schneider and Excoffier 1999). We examined these values separately for P. cinereus in each of the two contact zones (CZ1 and CZ2). Haplotype diversity in P. sherando was not high enough to permit analogous tests in this species.
Results
Plethodon sherando samples exhibited very low genetic diversity as compared to P. cinereus samples (Table 1). Across sites with no ambiguous bases, we recovered just 3 haplotypes with 2 variable nucleotide sites for P. sherando at both loci. Diversity was correspondingly low (h = 0.41, π = 0.0009). P. sherando samples from just the two contact zones yielded similar estimates of diversity (h = 0.56, π = 0.0010). In contrast, P. cinereus showed an order of magnitude greater nucleotide diversity within the same two contact zones (h = 0.633, π = 0.0096, with 5 haplotype and 14 variable sites (Table 1). Our six samples of P. hubrichti were intermediate between P. sherando and P. cinereus in sequence diversity (h = 0.60, π = 0.0051).
Genetic distance
Plethodon sherando COI and cyt-b haplotypes were highly divergent from P. cinereus haplotypes. Including only sites with no ambiguous bases, average pairwise percent sequence divergence was 16.7%, and genetic distance (GTR + Γ) between the lineages was 0.27 (±0.05). For comparison, the genetic distance between P. cinereus and P. hubrichti was 0.18 (±0.04).
Phylogenetic analysis
Phylogenies for both mtDNA segments based on maximum likelihood and Bayesian methods yielded identical topologies (Fig. 2). Reciprocal monophyly of animals that were morphologically P. sherando or P. cinereus was strongly supported (100% bootstrap support and 100% posterior probability for each clade). P. cinereus samples grouped into two clusters—one that corresponded with the high elevation sites (PC2 and CZ2) and one that corresponded to the low elevation sites (PC1 and CZ1). Because P. sherando showed very little sequence divergence across the range, there was no clustering based on collection site. Interestingly, P. cinereus grouped with P. hubrichti (rather than P. sherando), suggesting that P. sherando is a more distantly related lineage within the P. cinereus group.

Maximum likelihood and Bayesian phylogeny based on concatenated cyt-b and CO1 sequences with P. petraeus used as an outgroup. Numbers in parentheses are bootstrap support (proportion of bootstrap replicates) and posterior probabilities for each node. Nodes with support >0.98 are indicated with an “asterisk”. Site locations for samples (see Fig. 1) are given for each sample
In the expanded cyt-b phylogeny (Fig. 3), P. sherando was sister to the P. serratus rather than the P. cinereus. Although support for this node was only moderate (bootstrap probability = 0.78, posterior probability = 0.90), Highton (2004) reported the same putative relationship based on allozyme data. In any case, the phylogeny suggests that P. sherando is more distantly related to P. cinereus than are any of the other mountaintop species in the region (P. shenandoah, P. hubrichti).

Maximum likelihood and Bayesian phylogeny for cyt-b with P. petraeus used as an outgroup. Numbers in parentheses are bootstrap support (proportion of bootstrap replicates) and posterior probabilities for each node. Nodes with support >0.98 are indicated with an “asterisk”. Site locations for samples (see Fig. 1) are given for each sample. P. serratus and P. shenandoah samples are taken from GenBank
Time to most recent common ancestor
Plethodon sherando and Plethodon cinereus shared a common ancestor about 11.4 million years ago based on combined estimation from cyt-b and CO1 (95% HPD 9.0–13.8). P. sherando and P. serratus were estimated to have shared a common ancestor 6.0 million years (95% HDP 4.4–7.6) based on cyt-b sequences. Given the position of P. sherando on the phylogeny reported above (i.e. sister to P. serratus), these estimates are consistent with the fossil-calibrated phylogeny of Wiens et al. (2006).
Morphological analysis
Cluster analysis identified two clusters based on morphology for both reference site salamanders and for contact zone salamanders. For reference site samples (n = 109), BIC was substantially lower for a two-cluster model (equal volume, equal shape) as compared to a three-cluster model (ΔBIC = 14.1) or a single cluster (ΔBIC = 85.0). For contact zone samples (n = 79), BIC was also lowest for a two-cluster model as compared to three clusters (ΔBIC = 5.7) or a single cluster (ΔBIC = 39.9).
LDFA showed morphology at reference sites to be highly distinct between P. sherando and P. cinereus. Leave-one-out cross validation at reference sites yielded 100% classification success by species/site. Contact zone samples were, for the most part, also distinct. When individuals were classified based on morphology and validated based on mtDNA lineage, classification success was 98% for each lineage (Fig. 4). One P. sherando haplotype was classified as a morphological P. cinereus and one P. cinereus haplotype was classified as a morphological P. sherando. These 2 individuals were possibly hybrids. However, both individuals were juveniles, and lower posterior probabilities for other juveniles in the sample suggested that the classification model may have been less reliable for smaller salamanders. ANOVA indicated that, overall, salamanders in the contact zone were at least as distinct as salamanders at the reference sites. Interactions between lineage (P. sherando or P. cinereus) and site type (reference site or contact zone) were non-significant for both intercostal spaces/SVL (F1,109 = 0.69, P = 0.41) and for head width/SVL (F = 2.97, P = 0.08). The marginal result for head width (P = 0.08) is in the opposite direction than would be predicted with hybridization—lineages in the contact zone were more different than were lineages at the reference sites (Fig. 5). This result is plausibly consistent with findings of character displacement in head shape at contact zones between other pairs Plethodon (e.g. Adams 2004; Adams et al. 2007). Increased divergence in head width in contact zones was in fact statistically significant (F1,219 = 4.74, P = 0.03) when we increased our sample size by classifying salamanders by intercostal spaces instead of by mtDNA lineage.

Results for classification of contact zone salamanders with known mtDNA lineages based on morphological characters. All morphological classifications were consistent with mtDNA lineages except for the two indicated with arrows

Comparison of morphological differences between salamander species (as determined by mtDNA lineage) in contact zone areas versus reference sites. Tests for interactions between species and site type were used to ask whether salamanders were more similar in contact zones as compared to reference sites, as would occur with substantial introgression. Results are shown for: a intercostal spaces per unit SVL; b head width per unit SVL
Population history
In P. cinereus, sequence diversity was comparable between contact zone sites and reference sites (Table 1). Fu’s Fs was significantly negative in CZ2 (Fs = −1.32, P = 0.007), which is consistent with recent population expansion, though it was not significantly negative in CZ1 (Fs = −0.17, P = 0.079). Fs was not significantly different from zero for either of the reference sites PC1 or PC2 (Fs = −0.65, P = 0.11, and Fs = −0.53, P = 0.23). Harpending’s raggedness index, which indicates departure from unimodality in the mismatch distribution, only approached significance for the reference site PC2 (P = 0.07). The index was not significantly different from zero at any of the other three sites, indicating that a recent expansion model could not be rejected for these sites.
Discussion
Our results highlight the genetic uniqueness of the P. sherando lineage and the need to consider this species in conservation plans for the region. We found that morphological P. sherando were highly distinct from P. cinereus in their mtDNA sequences, with 17% sequence divergence across two mitochondrial regions. Our analysis of morphology in contact zones and reference sites further supported the distinction between these two taxa; all but two individuals were correctly assigned as P. sherando or P. cinereus on the basis of a few morphological characters, and there was little overall evidence of morphological intermediates detected in contact zones. The general concordance between mitochondrial genotype and morphology suggests that there has been at most a small amount of introgression between the lineages where they live in sympatry. Our phylogenetic analyses further supported this conclusion; P. sherando and P. cinereus morphotypes were reciprocally monophyletic using both maximum likelihood and Bayesian methods. These findings, combined with the very low genetic diversity found in P. sherando, suggest that P. sherando has a distinct evolutionary history and has probably undergone some period of isolation on or near the Big Levels ridgetop.
Conservation issues
Plethodon sherando’s extremely limited distribution (<80 km2) and its abrupt replacement by P. cinereus along its range boundaries create several potential conservation issues for this species. In general, anytime a species with a restricted distribution is surrounded by a more common congener, genetic swamping via introgression may be considered as a potential threat (Chan et al. 2006; Rhymer and Simberloff 1996). Moreover, many other Plethodon species are known to hybridize (Highton 1998, 1999) and hybridization may lead to substantial introgression in these species (Wiens et al. 2006; Weisrock et al. 2005). Indeed, we did find two misclassified individuals within the contact zone; these misclassifications could be due to measurement error, but they could represent true hybrids between P. sherando and P. cinereus. However, given the small number of these potential hybrids and the lack of any misclassifications at reference sites, we suggest that the threat of “genetic swamping” by P. cinereus does not appear to be an immediate threat to the persistence of P. sherando. Further studies at other contact zones between these two species would be helpful to ensure that introgression is not occurring extensively at other sites within P. sherando’s restricted range.
One of the most critical threats to mountaintop endemics like P. sherando may be climate change coupled with range expansion by competitors (Gilman et al. 2010). Milanovich et al. (2010) modeled salamander persistence in the southern Appalachians as a function of climate variables and showed that climate warming could lead to major range contractions. Their study did not include salamanders of the P. cinereus group; however, this group is also known to be sensitive to climate. For instance, Arif et al. (2007) found that the lower limit of P. hubrichti’s range appeared to be set by climate. For many mountaintop species, replacement by competing species with higher thermal tolerances is potentially a greater threat than just a change in the distribution of suitable habitats (Gilman et al. 2010). If P. cinereus outcompetes P. sherando at higher temperatures, expansion of P. cinereus into P. sherando’s range would likely follow climate warming. That said, the interaction of P. sherando with P. cinereus does not appear to reflect the traditional scenario of a mountaintop endemic surrounded by a lowland adapted species. Southwestern Virginia is near the southern limit of P. cinereus’ range, and P. cinereus’ complete range includes both high elevations within the Blue Ridge and colder regions as far north as central Quebec (Petranka 1998). Thus, it is not clear that warmer conditions would favor P. cinereus over P. sherando.
Evidence that P. cinereus is already expanding into the range of P. sherando was mixed. One contact zone site had a significantly negative value for Fu’s Fs (which can be indicative of recent population expansion) and one reference site had a value for Harpending’s raggedness index that approach significance (i.e. approached rejection of an expansion model). However, both references sites and contact zone sites had similar within-site genetic diversity, and most tests for expansion were non-significant. Ultimately, larger sample sizes will be necessary to understand the recent history of these contact zone populations.
Phylogeography
The phylogenetic analysis of P. sherando is interesting in that it suggests that, unlike P. shenandoah, P. sherando is not a sister species to P. cinereus. In fact, our analysis of cyt-b sequences suggests a potential affiliation between P. sherando and the P. serratus (Fig. 3). Although statistical support for this node was only moderate (79% ML boostrap support and 92% Bayesian posterior probability), Highton (2004) suggested a similar relationship based on independent allozyme data, and more recent nuclear data appear to confirm this affiliation (Highton, personal communication). P. serratus’ current range includes parts of Arkansas, Missouri, North Carolina Alabama, Georgia, Louisiana and Oklahoma (Petranka 1998). At the Northeastern limit of its range, P. serratus is only several hundred kilometers from the range of P. sherando. Given a divergence time of about 6 million years between the two lineages, it would not be surprising if P. sherando represents a lineage that became isolated in the Blue Ridge as the range of a P. serratus ancestor shifted southwards.
Study limitations
Our study has several important limitations. Most notably, we used concordance between mitochondrial genotypes and morphology to investigate the potential hybridization of the P. sherando and P. cinereus lineages. Although this approach has been used elsewhere (Babik et al. 2005; Carpenter et al. 2001; Szymura et al. 2000), clearly nuclear markers would have provided more direct evidence for or against hybridization. A second limitation is that we collected samples from only two contact zones, one at the lower limit of P. sherando’s elevational range and one near the upper limit. While we found no morphological evidence for hybridization at these sites, it is possible that hybridization was occurring at other sites where we did not collect. For example, morphological and allozymic data suggest hybridization between P. shenandoah and P. cinereus at one site but not at two others (Highton and Worthington 1967; Highton 1999). A third limitation is that we sampled only a portion of P. sherando’s range. Although we sampled opposite ends of the range, it is possible that we missed pockets of genetic diversity that would have changed our understanding of the P. sherando lineage. Finally, with only two mitochondrial regions, gene trees could differ substantially from true species trees (Degnan and Rosenberg 2009). That said, our results agree with the results of prior studies based on allozymes (Highton 2004) and forthcoming phylogenies of the Plethodon genus based on a large number of nuclear and mitochondrial markers (R. Highton, personal communication).
Conservation status of Plethodon sherando
Plethodon sherando currently lacks any special status within the U.S. or the State of Virginia because of its recent recognition and a lack of knowledge of its biology. In contrast, the other four mountaintop salamanders in the region all have some formal conservation status. The Shenandoah salamander (P. shenandoah) is listed as Federally Endangered and thus protected by the U.S. Endangered Species Act. The Cheat Mountain salamander (P. nettingi) is Federally Threatened and covered by a formal recovery plan. Both the Cow Knob salamander (P. punctatus) and the Peaks of Otter salamander (P. hubrichti) are listed federally as a “species at risk,” and are state-listed as a “species of concern.” For P. punctatus, this status means that most of its high-elevation habitat within National Forest is protected from logging (United States Forest Service 2011). For P. hubrichti, a primary habitat area is protected from logging and a secondary habitat area requires the consideration of management impacts on the species. Timber harvest within the secondary range of P. hubrichti is scheduled outside the normal surface activity periods for the species, and shelterwood and course woody debris are retained at sites where harvest occurs. Based on our findings, we suggest that P. sherando should merit a similar status to that of P. hubrichti. The range sizes of the two species are similar (~100 km2 for P. hubrichti versus ~80 km2 for P. sherando), and both species have ranges that consist primarily of high-elevation National Forest land. Moreover, both species’ distributions are entirely surrounded by P. cinereus at lower elevations, suggesting that competition, climate change, forest degradation, and interactions between these factors may be the greatest threats to long-term persistence. Based on these considerations, we believe that forest planning for the region should recognize the conservation needs of P. sherando as a spatially-restricted endemic species. This recognition could include the designation of P. sherando core habitat as a habitat conservation area, analogous to the habitat conservation areas that exist for both P. hubrichti and P. punctatus. In addition, as with P. hubrichti and P. punctatus, continued monitoring of range limits and restrictions on timber harvest within the species’ range should be important components of a long-term conservation strategy.
References
Adams DC (2004) Character displacement via aggressive interference in Appalachian salamanders. Ecology 85:2664–2670
Adams DC, West ME, Collyer ML (2007) Location-specific sympatric morphological divergence as a possible response to species interactions in West Virginia Plethodon salamander communities. J Anim Ecol 76:289–295
Anderson E, Maldonado-Ocampo J (2010) A regional perspective on the diversity and conservation of the tropical Andean fishes. Conserv Biol 25:30–39
Arif S, Wicknick JA, Adams DC (2007) Bioclimatic modeling, morphology, and behavior reveal alternative mechanisms regulating the distributions of two parapatric salamander species. Evol Ecol Res 9:843–854
Babik W, Branicki W, Crnobnja-Isailović J, Cogălniceanu D, Sas I, Olgun K, Poyarkov NA, Garcia-País M, Arntzen JW (2005) Phylogeography of two European newt species—discordance between mtDNA and morphology. Mol Ecol 14:2475–2491
Bensasson D, Zhang D-X, Hartl DL, Hewitt GM (2001) Mitochondrial pseudogenes: evolution’s misplaced witnesses. Trends Ecol Evol 16:314–321
Cabe P, Hanlon T, Aldrich M, Connors L, Marsh D (2007) Fine-scale population differentiation and gene flow in a terrestrial salamander (Plethodon cinereus) living in continuous habitat. Heredity 98:53–60
Carpenter D, Jung R, Sites J (2001) Conservation genetics of the endangered Shenandoah Salamander (Plethodon shenandoah, Plethodontidae). Anim Conserv 4:111–119
Chan C, Ballantyne K, Aikman H, Fastier D, Daugherty C, Chambers G (2006) Genetic analysis of interspecific hybridization in the world’s only Forbes’ parakeet (Cyanoramphus forbesi) natural population. Conserv Genet 7:493–506
Colliard C, Sicilia A, Turrisi G, Arculeo M, Perrin N, Stöck M (2010) Strong reproductive barriers in a narrow hybrid zone of west-mediterranean green toads (Bufo viridis subgroup) with Plio-Pleistocene divergence. BMC Evol Biol 10:232
Connors LM, Cabe PR (2003) Isolation of dinucleotide microsatellite loci from red-backed salamanders (Plethodon cinereus). Mol Ecol Notes 3:131–133
Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference, and the multispecies coalescent. Trends Ecol Evol 24:332–340
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis sampling trees. BMC Evol Biol 7:214
Duellman WE, Sweet SS (1999) Distribution patterns of amphibians in the Nearctic region of North America. In: Duellman WE (ed) Patterns of distribution of amphibians. Johns Hopkins Univ. Press, Baltimore, pp 31–109
Felsenstein J (1993) PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author. Department of Genetics, University of Washington, Seattle
Fraley C, Raftery AE (2006) MCLUST version 3 for R: normal mixture modeling and model-based clustering. Technical report. Department of Statistics, University of Washington, Seattle
Gilman SE, Urban MC, Tewksbury J, Gilchrist GW, Holt RD (2010) A framework for community interactions under climate change. Trends Ecol Evol 25:325–331
Harpending RC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Hastie T, Tibshirani R, Friedman JH (2001) Elements of statistical learning. Springer, New York
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W (2004) Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. PNAS 101:14812–14817
Hegde SG, Nason JD, Clegg JM, Ellstrand NC (2006) The evolution of California’s wild radish has resulted in the extinction of its progenitors. Evolution 60:1187–1197
Highton R (1995) Speciation in Eastern North American salamanders of the genus Plethodon. Annu Rev Ecol Syst 26:579–600
Highton R (1998) Frequency of hybrids between introduced and native populations of the salamander Plethodon jordani in their first generation of sympatry. Herpetologica 54:143–153
Highton R (1999) Geographic protein variation and speciation in the salamanders of the Plethodon cinereus group with the description of two new species. Herpetologica 55:43–90
Highton R (2004) A new species of woodland salamander of the Plethodon cinereus group from the Blue Ridge Mountains of Virginia. Jeffersoniana 14:1–22
Highton R, Worthington R (1967) A new salamander of the genus Plethodon from Virginia. Copeia 1967:617–626
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17:754–755
Jaeger RG, Forester DC (1993) Social behavior of plethodontid salamanders. Herpetologica 49:163–175
Johnson JA, Dunn PO, Bouzat JL (2007) Effects of recent population bottlenecks on reconstructing the demographic history of prairie chickens. Mol Ecol 16:2203–2222
Kleindorfer S, Chapman TW, Winkler H, Sulloway FJ (2006) Adaptive divergence in contiguous populations of Darwin’s Small Ground Finch (Geospiza fuliginosa). Evol Ecol Res 8:357–372
Knowles L (2001) Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshopprers. Mol Ecol 10:691–701
Kozak K, Wiens J (2006) Does niche conservatism promote speciation? A case study in North American salamanders. Evolution 60:2604–2621
Kozak KH, Wiens JJ (2010) Niche conservatism drives elevational diversity patterns in Appalachian salamanders. Am Nat 176:40–54
LaSorte F, Jetz W (2010) Projected range contractions of montane biodiversity under global warming. Proc R Soc B Biol Sci 277:3401–4310
Lawler JJ, Shafer SL, White D, Kareiva P, Maurer EP, Blaustein AR, Bartlein PJ (2009) Projected climate-induced faunal change in the Western Hemisphere. Ecology 90:588–597
Lovett JC (1998) Continuous change in Tanzanian moist forest tree communities with elevation. J Trop Ecol 14:719–722
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20:229–237
Mallet J, Beltrán M, Neukirchen W, Linares M (2007) Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol Biol 7:28
Marsh D, Page RB, Hanlon TJ, Corritone R, Little EE, Seifert DE, Cabe PR (2008) Effects of roads on patterns of genetic differentiation in red-backed salamanders, Plethodon cinereus. Conserv Genet 9:603–613
McCain CM (2005) Elevational gradients in diversity of small mammals. Ecology 86:366–372
Milanovich J, Peterman W, Nibbelink N, Maerz J (2010) Project loss of a salamander diversity hotspot as a consequence of project global climate change. PLoS ONE 5:e12189
Moritz C, Patton JL, Conroy CJ, Parra JL, White GC et al (2008) Impact of a century of climate change on small-mammal communities in Yosemite National Park, USA. Science 322:261–264
Mueller R (2006) Evolutionary rates, divergence dates, and the performance of mitochondrial genes in Bayesian phylogenetic analysis. Syst Biol 55:289–300
Myers N, Mittermeier R, Mittermeier C, Fonseca G, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858
Pauley T (2007) Revised notes on the range of the Cheat Mountain Salamander, Plethodon nettingi. Proc W Va Acad Sci 79:16–21
Petranka JW (1998) Salamanders of the United States and Canada. Smithsonian Institution Press, Washington
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Rhymer J, Simberloff D (1996) Extinction by hybridization and introgression. Annu Rev Ecol Syst 27:83–109
Rissler LJ, Smith WH (2010) Mapping amphibian contact zones and phylogeographic break hotspots across the United States. Mol Ecol 19:5404–5416
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Schneider S, Excoffier L (1999) Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Smith MA, Poyarkov NA, Hebert PDN (2008) CO1 DNA barcoding amphibians: take the chance, meet the challenge. Mol Ecol Resour 8:235–246
Steele C, Carstens BC, Storfer A, Sullivan J (2005) Testing hypotheses of speciation timing in Dicamptodon copei and Dicamptodon aterrimus (Caudata: Dicamptodontidae). Mol Phylogenet Evol 36:90–100
Szymura JM, Uzzell T, Spolsky C (2000) Mitochondrial DNA variation in the hybridizing fire-bellied toads, Bombina bombina and B. variegata. Mol Ecol 9:891–899
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Terborgh J (1977) Bird species diversity on an Andean elevational gradient. Ecology 58:1007–1019
United States Forest Service (2011) George Washington National Forest plan revision. http://www.fs.fed.us/. Accessed 11 November 2011
Weisrock D, Kozak H, Larson A (2005) Phylogeographic analysis of mitochondrial gene flow and introgression in the salamander, Plethodon shermani. Mol Ecol 14:1457–1472
Wiens J, Engstrom T, Chippindale P (2006) Rapid diversification, incomplete isolation, and the “speciation clock” in North American Salamanders (genus Plethodon): testing the hybrid swarm hypothesis of rapid radiation. Evolution 60:2585–2603
Wilson R, Gutiérrez D, Gutiérrez J, Monserrat V (2007) An elevational shift in butterfly species richness and composition accompanying recent climate change. Glob Change Biol 3:1873–1887
Xu T, Abbot R, Milne R, Mao K, Du F, Wu G, Ciren Z, Miehe G, Liu J (2010) Phylogeography and allopatric divergence of cypress species (Cupressus L.) in the Qinghai-Tibetan Plateau and adjacent regions. BMC Evol Biol 10:194
Acknowledgments
We thank Shane Ramee and members of the 2009 Field Herpetology class for help with collecting salamanders. Briana Gregory and Woodrow Friend assisted with DNA extractions and Jennifer Schieltz and Caroline Bovay contributed to the GIS analysis. We thank Richard Highton, Nadia Ayoub, and two anonymous reviewers for helpful comments on earlier drafts of this manuscript. This research was covered by Virginia state collection permit #33510 and IACUC animal care and use permit DM0109. Financial support for this study was provided by the U.S. Forest Service, a Howard Hughes Medical Institute award to Washington & Lee University under the Undergraduate Science Education Program, and an H. F. Lenfest grant to D Marsh.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bayer, C.S.O., Sackman, A.M., Bezold, K. et al. Conservation genetics of an endemic mountaintop salamander with an extremely limited range. Conserv Genet 13, 443–454 (2012). https://doi.org/10.1007/s10592-011-0297-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-011-0297-7