
Abstract
Tricyrtis ishiiana is a relic endemic plant taxon of the Convallariaceae that inhabits two nearby gorges in Kanagawa Prefecture, Japan. The distribution range and number of populations are thought to have been reduced to the present refugial populations during the Quaternary climatic oscillations. Because of its showy flowers, this plant has faced illegal removal from its natural habitats for horticultural use and has been designated a critically endangered species (class IA). In this study, we analyzed the genetic structure of the relict populations of T. ishiiana in order to contribute to the conservation strategies of the prefectural government. Our analyses of nine nuclear microsatellite loci detected high genetic diversity (HE = 0.704 and HO = 0.541) for the two populations. The two populations were slightly differentiated (RST = 0.032), accompanied by faint substructure across the populations (K = 3). In addition, each population exhibited spatial genetic structuring. The relatively low inbreeding coefficient for both populations together (FIS = 0.233) and each population separately (FIS = 0.217–0.246) may be attributable to crossing among descendants within a population along with occasional gene flow between the populations. These results suggested that the extant populations have not experienced a severe bottleneck. The two extant populations were genetically differentiated at a very low level, accompanied by occasional pollen flow via pollinators and/or seed dispersal by gravity in the mountainous environment. Occasional gene exchange between the populations has allowed T. ishiiana to harbor high genetic diversity despite being a relic plant confined to two small refugial populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Tricyrtis ishiiana (Kitag. et T. Okuyama) Ohwi et Okuyama is a perennial herb of the Convallariaceae that is confined to two gorges in the Tanzawa Mountains in Kanagawa Prefecture, Japan (Fig. 1). This species inhabits precipices of gorges in the cool-temperate forest at elevations ranging from ~800 to 1000 m. The large (approximately 5 cm in length) showy flowers are yellow and bell-shaped, and are pollinated by bumblebees (Takahashi 1993). This species is vulnerable because of its limited population number and illegal removal from its natural habitats for horticultural use. It has been designated a critically endangered species (class IA) based on IUCN criteria (Japan Society of Plant Taxonomists 1993; Ministry of the Environment of Japan 2000).

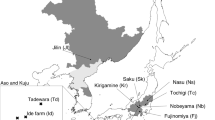
Locations of the study sites. a Location of the study sites within the Japanese islands. Closed circles with arrows indicate the locations of the two T. ishiiana populations. b Map of the Tanzawa Mountains, showing river systems and the locations of the two T. ishiiana populations examined (enclosed by squares)
Increased outcrossing is a result of the protandry of T. ishiiana flowers (Takahashi 1993); however, crossing between kin-individuals may result in lower genetic diversity of the relic population. In addition, this plant extends horizontal rhizomes and propagates clonally. Consequently, the genetic diversity of the two confined populations may decrease as a result of inbreeding, clonal propagation, and a bottleneck effect for population establishment. Evaluation of the population genetic diversity and population genetic structure would provide basic information for the conservation of the remaining populations.
In addition, estimation of the gene flow between the two populations would provide conservation strategies, delimiting whether the two populations should be protected individually or inclusively. The two habitat gorges are approximately 640 m apart (Fig. 1), and pollen flow between the two populations may be mediated within this distance by bumblebees. In addition, seed dispersal is believed to occur by gravity or strong wind, based on the relatively small seed size (3–4 mm in diameter) and the presence of a narrow wing. Thus, there are a few opportunities for seed dispersal between the populations. On the other hand, this plant grows on precipices of gorges, and seed and pollen flows may tend to remain confined within a gorge. In this case, genetic differentiation would be high between the populations.
Relic endemic species confined to small refugial populations may harbor either low or high genetic diversity. The extant populations were reduced and retreated to their distribution range during the Quaternary climatic oscillations. The small distribution range and low number of populations suggests that the species has a diminished genetic diversity due to a bottleneck effect. In general, narrowly restricted species tend to harbor lower levels of a genetic diversity than widespread species (e.g., Sherman-Broyles et al. 1992; Linhart and Permoli 1993; Edwards and Wyatt 1994; Purdy and Bayer 1995a, b, 1996; Godt et al. 1997). On the other hand, phylogeographic examinations of Quaternary climatic oscillations and range shifts in biota have revealed that persisting populations tend to exhibit higher genetic diversity and/or haplotype diversity than those of recolonized populations (e.g., Lumaret et al. 2002; Palmé and Vendramin 2002; Petit et al. 2002, 2003; Schönswetter et al. 2005; Ikeda et al. 2006). Although T. ishiiana has only two neighboring populations with small ranges, these populations (or at least, one of the two populations) may harbor high genetic diversity originating from the ancestral population. In the latter case, a serious bottleneck effect might not have occurred at the time of population establishment and/or division into the two gorges.
The Kanagawa prefectural government has recently been concerned with the genetic signature of T. ishiiana in terms of conservation strategy planning. In this study, we analyzed the genetic structure of the two relict populations of this plant located in nearby gorges. Knowledge of the gene flow mediated by pollen and seed dispersal could provide new insights into the population dynamics of the refugial populations and would contribute to the conservation of these relict populations. We examined the population genetic structure based on highly polymorphic nuclear microsatellite loci, which allow the determination of fine-scale genetic structure.
Materials and methods
Sampling
Tricyrtis ishiiana is a deciduous perennial herb that grows in shaded precipices on the slopes of two gorges: Taraigoyasawa Gorge (population TA) and Sedonosawa Gorge (SE) in the Tanzawa Mountains of Kanagawa Prefecture. No specimens have been found in the forest understory or in sunny locations. The two populations are located at the upper ends of the gorges, ranging about 350 and 410 m along the mountain stream in TA and SE, respectively. The two populations are separated by a mountain ridge and approach most closely at a distance of approximately 640 m (Fig. 1). The large showy flowers have a corolla approximately 5 cm in length and are yellow and bell-shaped, and visited by bumblebees (Takahashi 1993). Seed dispersal is thought to occur via gravity, given the small seed size (~2–3 mm across) and the narrowly winged appendages. Seeds are not produced by selfing under cultivated conditions, and outcrossing is assumed.
Fresh leaves of mature individuals were sampled from the two populations (Fig. 1). This plant is usually lithophytic, i.e., grows in or on rocks; thus, we could not observe its root system without tearing plants and moss from rocks. Therefore, plants located at least several meters apart were selected, to avoid sampling kin-individuals and/or clonal ramets. Details of the sampling localities and the number of individuals used for the analyses are shown in Table 1. Population TA was larger, with more than 107 available ramets growing on a cliff facing the gorge; the smaller SE population had 62 samples available for analysis. In total, 169 samples were used for DNA analysis.
DNA extraction, PCR amplification, and microsatellite genotyping
Fresh leaf material was frozen in liquid nitrogen and then ground into a fine powder. After the polysaccharides were removed from the powder using HEPES buffer (pH 8.0; Setoguchi and Ohba 1995), genomic DNA was extracted using the cetyltrimethyl ammonium bromide (CTAB) method (Doyle and Doyle 1990). The extracted DNA was dissolved in 100 μl of TE buffer and used for PCR. The genotypes of 169 individuals were determined using nine nuclear simple sequence repeat (nSSR) markers developed for this study (TI-1, TI-2, TI-3, TI-4, TI-5, TI-6, TI-7, TI-8, TI-9; Setoguchi et al. 2009). The primers used are listed in Table 2.
PCR was performed in a 6-μl reaction volume containing 40–60 ng of genomic DNA, 1× Multiplex PCR Master Mix (Qiagen), and 0.2 μM of each primer. Samples were amplified in a DNA thermal cycler (Takara Bio) programmed for denaturation at 95°C for 15 min, followed by 30 cycles at 94°C for 30 s, 58°C for 90 s, and 72°C for 60 s, and a final 30-min extension at 60°C. The amplified products were loaded onto an ABI 3100 autosequencer (Applied Biosystems) using POP6 polymer and a 50-cm capillary array, and their sizes were determined using Genemapper (Applied Biosystems). The use of the polymer and capillary array enabled us to determine relatively short fragments (<100 bp).
Data analyses
The number of alleles, allelic richness (El Mousadik and Petit 1996), observed heterozygosity (H O), gene diversity (H E; Nei 1987), and fixation index (F IS = 1 − H O/H E) were calculated for each locus and each population (TA: 107 samples; SE: 62 samples; total number of samples: 169) using fstat ver. 2.9.3.2 (Goudet 1995). Departures from Hardy–Weinberg equilibrium (HWE) at each locus and the linkage disequilibrium between loci were tested using fstat (alleles were randomized 1,000 times over all samples). These parameters were calculated for 107 and 62 individuals in the TA and SE populations, respectively.
Genetic differentiation among populations (R ST; Weir and Cockerham 1984; Slatkin 1995; Rousset 1996) were estimated using fstat. R ST is the coefficient of genetic differentiation among populations assuming the stepwise mutation model. Significant differences among these parameters at each locus were tested by the log-likelihood (G)-based exact test (Goudet et al. 1996) using a Monte Carlo Markov chain (MCMC) method.
Population differentiation based on pairwise R ST between populations was estimated using arlequin version 3.1 (Excoffier et al. 2005).
Relatedness coefficients (r-values) between all possible pairs of individuals were estimated using KINSHIP 1.3.1 (Queller and Goodnight 1989). The calculation is based on the frequency of alleles in the population. This measure of relatedness ranges from −1 to +1, where a positive value indicates that two individuals share more identical alleles than expected by chance.
In addition, we used a Bayesian clustering approach implemented in structure ver. 2.2 (Pritchard et al. 2000) to infer population structure. This approach assumes HWE and attempts to find population groupings that are not in linkage or Hardy–Weinberg disequilibrium. To quantify the variation in likelihood for each number of clusters (K), we performed a series of 20 independent runs for each value of K, ranging from 1 to 9. As recommended in the software manual, we assumed an admixture model of ancestral populations (Falush et al. 2003) with a correlated model of allele frequencies using 1 × 105 burn-in time periods and 5 × 106 MCMC iterations. Previous studies have shown that in many cases, the posterior probability for a given K increases slightly, even after the real K is reached. Therefore, we used the ad hoc statistic of Evanno et al. (2005), ∆K, to determine the true value of K.
The genetic distance and model-based clustering methods described above estimate recent gene flow among populations. In addition, the method described by Wilson and Rannala (2003) estimates the amount and direction of recent (i.e., over the past few generations) gene flow. We estimated recent gene flow using the program BayesAss ver. 1.3, which estimates recent migration rates between all pairs of populations, allele frequencies, and inbreeding coefficients for each population. These analyses were performed by identifying individuals as immigrants or as descendents of recent immigrants from the observed temporary disequilibrium of multi-locus genotypic frequencies. The parameters (migration rate, allele frequency, and inbreeding coefficient) were estimated numerically with a MCMC simulation by inferring the estimated posterior probability. To estimate the posterior probability distribution of the parameters, the program was run with 999,999 burn-in periods and 3 × 106 total iterations. Five independent runs were conducted, and the mean values were compared among populations. We conducted the analysis for two populations, downstream and upstream, from each of the two gorges (TA and SE), thus examining four populations in total.
The patterns of spatial genetic structure described as isolation-by-distance (IBD) model (Wright 1943) were evaluated according to Rousset (1997). A Mantel test with 10,000 random permutations was performed with the matrix of pairwise genetic differentiation between subpopulations using R ST/(1 − R ST) and a matrix of the ln (geographic distance) using the program GENALEX version 6.1 (Peakall and Smouse 2006). A spatial autocorrelation analysis was performed with SPAGEDI version 1.2 (Hardy and Vekemans 2002). This analysis used R ST and evaluated four to seven spatial distance classes with similar sample sizes. The 95% confidence intervals were estimated for each distance class using 10,000 random permutations.
To detect recent bottlenecks due to reductions in effective population size, the observed gene diversity was compared with equilibrium gene diversity given the observed number of alleles (Watterson 1978, 1986) using bottleneck 1.2.02 (Piry et al. 1999). Two models for locus evolution, the infinite allele model (IAM; Maruyama and Fuerst 1985) and stepwise mutation model (SMM; Cornuet and Luikart 1996), were used for the analyses, with a Sign test (Cornuet and Luikart 1996) and a Bayesian Wilcoxon signed-rank test (Luikart et al. 1998).
Results
Genetic diversity
Characteristics of the nine nSSR loci examined are shown in Table 2. In total, 137 alleles for the nine nSSR loci were detected. These alleles distinguished all samples as different genets, implying that no clone was found among samples. Significant deviation (P < 0.05) from HWE was detected in five of the nine loci, possibly due to depressed gene flow between the TA and SE populations and low genetic diversity of the loci (in particular TI-6 and TI-7). High levels of genetic diversity were consistently observed in each population, as indicated by H E estimates of 0.706 (population TA) and 0.702 (SE), with an average of 0.704 (Table 3). H O was relatively low compared to H E and ranged from 0.529 (SE) to 0.553 (TA), with an average of 0.541. Consequently, F IS calculated for each population ranged from 0.217 (TA) to 0.246 (SE), with an average of 0.232. No significant linkage disequilibrium between loci across the two populations was observed, and all loci were used for further analyses. The degrees of relatedness represented by the r-values were −0.004 ± 0.003 (mean ± standard error; population TA) and −0.002 ± 0.006 (population SE; Table 3). In addition, subpopulations g and h within the TA population (as large subpopulations comprising n > 20) harbored relatedness coefficients of −0.015 ± 0.014 and 0.018 ± 0.016, respectively.
Genetic differentiation and gene flow between populations
RST between the two populations was 0.032, whereas most (~93%) of the genetic variation was explained by differentiation within the populations (Table 4). Genetic differentiation was significant (P < 0.001) among populations at all loci (Goudet et al. 1996).
The structure analysis detected three appropriate clusters (K). Individual assignments into the three clusters were consistent in the 20 replicate runs. Population SE belonged to one cluster, and population TA was split into two clusters (Fig. 2). In addition, the genetic composition determined by the Bayesian clustering is indicated by pie charts for each population (Fig. 3). Populations TA and SE were clearly distinguished, whereas population substructure and special distribution of the genetic composition were slightly recognizable within each population: upstream and downstream populations tended to be occupied by different clusters of genotypes (e.g., dark and light gray, respectively, in TA; Fig. 3). The spatial genetic structure as isolation by distance was detected at different spatial scales (Mantel test; P < 0.05): at scales of the whole distribution range (Fig. 4, W), within TA (Fig. 4, TA), and within SE (Fig. 4, SE). The geographic pattern of genetic variation was analyzed by the spatial autocorrelation analysis (Fig. 5). Within the TA population, R ST value between subpopulations separated by more than about 370 m was significantly higher than the expected value (Fig. 5, TA). The SE population also harbored a similar geographic structure; a higher average genetic distance (R ST) was estimated as being more than about 450 m separating the subpopulations (Fig. 5, SE). At the whole distribution range level, subpopulations between about 200 m and about 1000 m exhibited spatial autocorrelation in the average genetic distance (Fig. 5, W).

Results of structure analysis. a Distribution of likelihood values, Ln(K). All estimated values from 20 replicates in each K were plotted in Ln(K). b Distribution of the model parameter (∆K). The estimated values based on the 20 replicates are shown in ∆(K). c Individual assignment by structure analysis: the number of clusters (K) was three. Bold lines within the squares distinguish populations. Abbreviations for each population name are shown under the bars

Location and genetic structure of the subpopulations of T. ishiiana. The pie chart indicates the genetic composition of clusters determined by Bayesian clustering (structure analysis). The size of the chart corresponds to the number of individuals. Codes for the subpopulations correspond to those in Table 1

Relationship among the matrix of pairwise differentiation described as R ST/(1 − R ST) and the matrix of the natural logarithm of geographical distance among individuals of T. ishiiana for the whole distribution range (W), Taraigoyasawa Gorge (TA), and Sedonosawa Gorge (SE)

Average genetic distance (R ST) for special distance classes for seven classes in the whole distribution range (W), four classes in Taraigoyasawa Gorge (TA), and four classes in Sedonosawa Gorge (SE). Broken lines indicate the boundaries of the 95% confidence intervals estimated by permutation
Estimates of recent migration rates between populations (up to the second generation of migrants) are shown in Table 5a. The 95% confidence interval is also presented in parentheses. Recent gene flow among populations was calculated as 0.188 from SE to TA and 0.198 from TA to SE, of which the direction of gene flow could not be evaluated (ANOVA, P > 0.05). Analyses for gene flow among four subpopulations are presented in Table 5b. Gene flow from upstream to downstream (mean, 0.035) was significantly higher than that from downstream to upstream (mean, 0.008) in TA (ANOVA, P < 0.05). In another gorge (SE), gene flow from upstream to downstream was relatively high (mean, 0.129) but this value was nonsignificant based on 95% confidence intervals and also the manual of the BayesAss program. This value was higher than that of the opposite direction (mean, 0.050), but the difference again was not significant (ANOVA, P > 0.05).
The results of tests for mutation-drift equilibria are shown in Table 6. The Sign test under the IAM showed no evidence of a recent bottleneck in either the whole population or each individual population (P > 0.05), whereas significant bottleneck effects were detected over all populations and for each population under the SMM. The Wilcoxon test estimated a bottleneck effect in one population (TA) and in over all populations, but not SE. Thus, three of the four analyses detected a bottleneck effect in the TA population, and a bottleneck effect in the SE population was detected by one of the four analyses.
Discussion
Genetic diversity of relict populations
These results indicated that both populations of T. ishiiana in the two gorges harbor high genetic diversity (H O = 0.541 and H E = 0.704), although both populations are very small and confined to two narrow gorges (distribution range is about 400 m in each gorge). These values are almost equal to those of other Tricyrtis species in Japan, which are outcrossing and narrowly restricted ones: T. perfoliata (distribution is confined to one mountain): H E = 0.693; T. ohsumiensis (restricted to a few mountains in southern Kyushu): H E = 0.788; and T. flava (restricted to southeastern Kyushu): H E = 0.747 and (Takahashi et al., in press). In terms of relict populations, isolated populations of Kirengeshoma palmata (outcrossing perennial herb of the Hydrangeaceae) in Japan harbor genetic diversity as I (Shannon’s diversity index) = 0.261 and H E = 0.175 (ISSR analysis: Qiu et al. 2009). Although these data are based on different locus or molecular markers, the genetic diversity of T. ishiiana is high as well as congeners and other relict endemic plant, possibly due to its population history (range shifts and population size change) during the Quaternary climatic oscillations. Many phylogeographic studies have revealed that refugial populations tend to have higher genetic and/or haplotype diversity than those of recolonized populations (e.g., Lumaret et al. 2002; Palmé and Vendramin 2002; Petit et al. 2002, 2003; Schönswetter et al. 2005; Ikeda et al. 2006). Narrowly restricted populations of T. ishiiana harbor high genetic diversity, but this plant has only two relict populations without any recolonized ones. Therefore, we should be careful as to identifying the extant populations as having been in “refugia” during the Quaternary.
The high genetic diversity in T. ishiiana appears to contradict the result of the bottleneck analyses, which showed evidence of reductions in effective population size. The entire T. ishiiana population would have certainly decreased in size following range contraction during the postglacial period and ultimately reached safety in the relict populations. Therefore, the larger ancestral populations harboring genetic diversity might have migrated to the present locality and reduced the distribution range. The retreated populations had experienced population size reduction, but they have kept the aforementioned genetic diversity. Thus, the present populations can be viewed as populations that survived during the postglacial warm periods and maintained genetic diversity.
Genetic structure of populations in the two gorges
This study also indicated that the two extant populations of T. ishiiana are genetically differentiated at a very low level (R ST = 0.032), whereas a faint substructure across the populations was suggested by the structure analysis (K = 3). The SE population was composed of a single cluster of genotypes in the structure analysis, and the TA population included two clusters of genotypes. The presence of potential differentiation between the two populations implies the presence of a weak barrier to gene flow via pollen flow and seed dispersal between the two populations. The TA and SE populations are approximately 640 m apart as the minimum distance, partitioned by a mountain ridge (Fig. 1). Therefore, the topography might have restricted gene flow and sculpted the population substructure. On the other hand, the Bayesian assessment determined by the BayesAss program estimated recent migration between the two populations. These results along with a very low R ST value suggested that the two extant populations were genetically differentiated at a very low level, accompanied by occasional pollen flow via pollinators and/or seed dispersal by gravity in the mountainous environment.
On the other hand, we were able to identify spatial genetic structuring within each gorge. The TA and SE populations harbored spatial genetic structuring within less distance (less than about 370 m and less than about 450 m, respectively), implying the presence of gene flow within a gorge, possibly attributable to pollen flow and/or seed flow. The significant but slight gene flow from upstream to downstream in the TA gorge may suggest the presence of occasional gene flow by water along the river, but we cannot interpret this result in terms of pollen and seed dispersal based on our data. The spatial genetic structure of Tricyrtis seeds is presumed to be dispersed by gravity or strong winds, without adaptations for water dispersal, and pollen is moved by pollinator insects such as bumblebees. Thus, the spatial genetic structuring of T. ishiiana could represent a signature of pollen and seed dispersal for this plant.
Tricyrtis ishiiana is a typical relic endemic species, confined to a small distribution range of cool riparian crags. Based on the high genetic diversity within the extant descendents, the ancestral populations would have to have been large enough to harbor high genetic diversity. During the subsequent postglacial range contraction, the retreating populations might have maintained a population size large enough to retain genetic diversity without experiencing a bottleneck effect. The high genetic diversity of T. ishiiana populations may imply the genetic signature of relic populations of narrowly endemic plants.
Conservation perspectives
This study provides genetic information on refugial populations of T. ishiiana. Maki et al. (1999) compared fixation indices for four species of Tricyrtis in terms of mating systems. Adichogamous and putative selfing T. nana showed high values of Wright’s fixation index (f = 0.534–0.780), whereas those of protandrous and putative outcrossing species varied: f = 0.021–0.243 for widespread T. flava, f = 0.040 for narrowly endemic T. perfoliata, and f = 0.249 for narrowly endemic T. ohsumiensis. Later, Takahashi et al. (in press) reported Wright’s fixation index, F IS = 0.661, for T. nana (no data for other species). Thus, our result (F IS = 0.232) can be compared to those of protandrous and outcrossing congeners, and agree with the breeding system described by Takahashi (1993). Accordingly, the fairly high heterozygosity of T. ishiiana (H O = 0.541, H E = 0.704) would be partly attributable to breeding system.
Low genetic differentiation between the populations (R ST = 0.032) suggested that the entire habitat (including both populations) should be protected as one unit, without treating the two populations as separate. Nevertheless, transplantation and/or artificial seed movement should be avoided because the two populations harbor their inherent clusters of genotype, as detected in the Bayesian clustering analysis.
This plant should be considered for protection, as it reflects Quaternary climatic oscillation. Considering the presence of spatial genetic structure in extant populations, we recommend to protect each population (and subpopulation) without transplantation among/within populations. The genetic diversity and genetic signature of each population and subpopulations should be protected from illegal removal for horticultural use.
References
Cornuet JM, Luikart G (1996) Description and evaluation of two tests for detecting recent bottlenecks. Genetics 144:2001–2014
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from plant tissue. Focus 12:13–15
Edwards AL, Wyatt R (1994) Population genetics of the rare Asclepias texana and its widespread sister species, A. perennis. Syst Bot 19:291–307
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Godt MJW, Walker J, Hamrick JL (1997) Genetic diversity in the endangered lily Harperocallis flava and a close relative, Tofieldia racemosa. Conser Biol 11:361–366
Goudet J (1995) Fstat version 1.2: a computer program to calculate F statistics. J Hered 86:485–486
Goudet J, Raymond M, De-Meeus T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Hardy OJ, Vekemans X (2002) SPAGEDI: a versatile computer program to analyze spatial genetic structure at the individual or population level. Mol Ecol Notes 2:618–620
Ikeda H, Senni K, Fujii N, Setoguchi H (2006) Refugia of Potentilla matsumurae (Rosaceae) located at high mountains in the Japanese archipelago. Mol Ecol 15:3731–3740
Japan Society of Plant Taxonomists (1993) Red Data Book: Japanese endangered plants. Nousonbunka-sha, Tokyo (in Japanese)
Linhart YB, Permoli AC (1993) Genetic variation in Altes acaulis and its relative, the narrow endemic A. humilis (Apiaceae). Am J Bot 80:598–605
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89:238–247
Lumaret R, Mir C, Michaud H, Raynal V (2002) Phylogeograpical variation of chloroplast DNA in holm oak (Quercus ilex L.). Mol Ecol 11:2327–2336
Maki Y, Morita H, Oiki S, Takahashi H (1999) The effect of geographic range and dichogamy on genetic variability and population genetic structure in Tricyrtis section Flavae (Liliaceae). Am J Bot 86:287–292
Maruyama T, Fuerst PA (1985) Population bottlenecks and non equilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 111:675–678
Ministry of the Environment of Japan (2000) Threatened wildlife of Japan, Red Data Book, vol 8, 2nd ed (vascular plants). Japan Wildlife Research Center, Tokyo (in Japanese)
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Palmé AE, Vendramin GG (2002) Chloroplast DNA variation, postglacial recolonization and hybridization in hazel, Corylus avellana. Mol Ecol 11:1769–1779
Peakall R, Smouse PE (2006) Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam B, Deans JD, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Jensen JS, König AO, Lowe AJ, Madsen SF, Mátyás G, Munro RC, Popescu F, Slade D, Tabbener H, de Vries SGM, Ziegenhagen B, de Beaulieu JL, Kremer A (2002) Identification of refugia and post-glacial colonization routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manag 156:49–74
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Muller-Starck GM, Demesure-Musch B, Palme A, Martin JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Purdy BG, Bayer RJ (1995a) Allozyme variation in the Athabasca sand dune endemic, Salix silicicola, and the closely related widespread species, S. alaxensis. Syst Bot 20:179–190
Purdy BG, Bayer RJ (1995b) Genetic diversity in the tetraploid sand dune endemic Deshampia mackenzieana and its widespread progenitor D. cespitosa (Poaceae). Am J Bot 82:121–130
Purdy BG, Bayer RJ (1996) Genetic variation in populations of the endemic Achillea millefolium ssp. megacephala from the Athabasca sand dunes and the widespread ssp. lanulosa in western North America. Can J Bot 74:1138–1146
Qiu YX, Sun Y, Zhang XP, Lee J, Fu CX, Comes HP (2009) Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to quaternary climate change and landbridge configurations. New Phytol 183:480–495
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evol Int J Org Evol 43:258–275
Rousset F (1996) Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics 142:1357–1362
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Schönswetter P, Stehlik I, Holderegger R, Tribsch A (2005) Molecular evidence for glacial refugia of mountain plants in the Europe Alps. Mol Ecol 14:3547–3555
Setoguchi H, Ohba H (1995) Phylogenetic relationships in Crossostylis inferred from restriction site variation of chloroplast DNA. J Plant Res 108:87–92
Setoguchi H, Mitsui Y, Ikeda H, Nomura N, Tamura A (2009) Development and characterization of microsatellite loci in the endangered Tricyrtis ishiiana (Convallariaceae), a local endemic plant in Japan. Conserv Genet 10:705–707
Sherman-Broyles SL, Gibson JP, Hamrick JL, Bucher MA, Gibson MJ (1992) Compariosn of allozyme diversity among rare and widespread Rhus species. Syst Bot 17:551–559
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequency. Genetics 139:457–462
Takahashi H (1993) Floral biology of Tricyrtis macranthopsis Masamune and T. ishiiana (Kitagawa et T. Koyama) Ohwi et Okuyama (Liliaceae). Acta Phytotaxonomica et Geobotanica 44:141–150
Takahashi Y, Takahashi H, Maki Y Comparison of genetic variation and differentiation using microsatellite markers among three rare threatened and one widespread toad lily species of Tricyrtis section Flavae (Convallariaceae) in Japan. Plant Species Biol (in press)
Watterson GA (1978) The homozygosity test of neutrality. Genetics 88:405–417
Watterson GA (1986) The homozygosity test after a change in population size. Genetics 112:899–907
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wright S (1943) Isolation by distance. Genetics 28:114–138
Acknowledgments
We are grateful to Ms. Akiko Sakai (Kanagawa Prefecture Natural Environment Conservation Center), and Asuka Noda, Kanako Sugahara and Tomomi Dan (Kyoto University) for their cooperation in collecting materials and extracting DNA, respectively. Prof. Masayuki Maki (Tohoku University) kindly showed us his unpublished data (currently in press). This study was supported by a grant for Conservation Genetics for Endangered Plants from Kanagawa Prefecture Natural Environment Conservation Center.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Setoguchi, H., Mitsui, Y., Ikeda, H. et al. Genetic structure of the critically endangered plant Tricyrtis ishiiana (Convallariaceae) in relict populations of Japan. Conserv Genet 12, 491–501 (2011). https://doi.org/10.1007/s10592-010-0156-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0156-y