
Abstract
Dramatic changes in environmental conditions or community composition may impose severe selective pressures on resident populations. These changes in the selective regime can lead to demographic bottlenecks or local extinction. The consequence of demographic contraction is often a reduction of standing genetic variation. Since the level of adaptive genetic variation in populations plays an important role in persistence and adaptive response, understanding genetic resilience and the time course for re-establishment of genetic diversity following demographic perturbations is a critical component of assessing the consequences of changing environments. The introduction of nonnative fish into historically fishless lakes is a particularly dramatic environmental change frequently contributing to demographic bottlenecks and local extinction of native populations. We examine the quantitative- and molecular-genetic recovery of two alpine populations of the zooplankton Daphnia melanica from the Sierra Nevada, California, USA. These populations were extirpated by introduced salmonids and subsequently re-established following the experimental removal of nonnative fish. We obtained data for nuclear and mitochondrial markers and conducted a common-garden experiment to assess the levels of molecular- and quantitative-genetic variation following experimental fish removal. Reestablished D. melanica populations attained levels of nuclear genetic diversity only slightly lower than surrounding fishless populations in the first year following fish removal and substantial mitochondrial and quantitative-genetic diversity within 8 years. This high level of genetic resilience was likely facilitated by multiple sources of genetic variation, including immigration from neighboring populations and hatching from a local reservoir of diapausing eggs. Our results highlight the genetic resilience of taxa with reservoirs of genetic variation in seed or egg banks.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Demographic concerns have historically been the primary consideration guiding conservation management strategies. Increasingly, the significance of maintaining high levels of genetic diversity for adaptive traits has become more widely appreciated (Frankham et al. 2002; Kohn et al. 2006; Bonin et al. 2007). Both demographic and genetic recovery are important following demographic collapse. Rapid population growth alleviates stochastic threats to population persistence while genetic diversity provides material for adaptive response to subsequent changes in the environment. The importance of genetic variation is highlighted by consideration of the deleterious impacts of inbreeding depression in small populations (Tallmon et al. 2004), recent theoretical and empirical treatments of the relationship between adaptation and population persistence in changing environments (Gomulkiewicz and Holt 1995; Orr and Unckless 2008; Bell and Gonzalez 2009), and the realization that many populations undergo rapid adaptive responses to changes in their environment (Thompson 1998; Hendry and Kinnison 1999; Cousyn et al. 2001; Fisk et al. 2007).
One reason genetic diversity has not always been a management priority is that genetic recovery often occurs on time-scales that far exceed that of pure demographic recovery. Northern elephant seals provide a classic demonstration of the disparity between recovery of genetic variation and population expansion. Following a bottleneck in the 1880s that reduced the population to as few as 20 individuals, northern elephant seals currently number around 175,000 (Weber et al. 2000). However, decades later no detectable genetic diversity for 20 allozymes (Bonnell and Selander 1974), and only two mitochondrial variants (Hoelzel et al. 1993) were found in the extant population. Thus, while these populations are buffered against extinction risk from environmental and demographic stochasticity, they remain prone to extinction because of their compromised adaptive potential to selective challenges such as novel pathogens and environmental change.
Species introductions or invasive species entering naïve communities often contribute to demographic reductions in the population size of native species and a corresponding loss of genetic variation (Sakai et al. 2001). This pattern is the frequent outcome following the introduction of nonnative fish into historically fishless lakes in alpine ecosystems (Parker et al. 1996; Bradford et al. 1998; Donald et al. 2001; Knapp et al. 2001a; Schindler and Parker 2002). These introductions have repeatedly led to population bottlenecks and local extinction of native amphibian, macroinvertebrate, and zooplankton populations (Bradford et al. 1998; Knapp and Matthews 2000; Knapp et al. 2001b). Accordingly, several species, particularly amphibians whose populations have been reduced to less than 10% of historical sizes in some cases (Vredenburg et al. 2006), are now high priority targets for conservation efforts.
When fish are removed from freshwater lakes, native populations often undergo periods of demographic expansion. Experimental removal of fish from lakes has resulted in the recovery of native populations of the mountain yellow-legged frog, Rana muscosa, in the Sierra Nevada, California (Vredenburg 2004; Knapp et al. 2007) and northwestern salamander, Ambystoma gracile, in the northern Cascade Mountains of Washington (Hoffman et al. 2004). Zooplankton populations that produce dormant resting eggs are particularly suited for rapid demographic recovery following fish removal. Experimental fish removal facilitated the recolonization and establishment of previously extinct Daphnia and copepod populations in lakes of the Sierra Nevada (Knapp et al. 2001b; Sarnelle and Knapp 2004; Knapp and Sarnelle 2008) and the Daphnia population in Bighorn Lake in Alberta, Canada (Parker et al. 2001). Thus, lakes in which fish have been experimentally removed provide a unique opportunity for simultaneous investigation of the dynamics of demographic and genetic recovery in natural populations following population bottlenecks and local extinction.
The goal of this study is to estimate levels of neutral and quantitative genetic variation in two D. melanica populations (“restoration populations”) from the Sierra Nevada in which the planktonic population was extirpated prior to the experimental removal of nonnative fish. To achieve our goal we collected allelic data for nuclear loci (microsatellites) and sequence data for mitochondrial protein coding genes, which provide coarse estimates of overall genomic diversity. We also conducted a common-garden experiment to estimate levels of quantitative genetic variation for ecologically relevant traits. We compared the levels of neutral and quantitative genetic variation in restoration populations 1 and 8 years after fish removal with estimates of genetic variation in D. melanica populations with no history of fish introductions (“fishless populations”). This study builds on previously described demographic response to fish removal in these populations (Sarnelle and Knapp 2004).
Materials and methods
Study populations
Daphnia melanica used in this study were collected from six permanent lakes in the central Sierra Nevada in Fresno and Tuolumne counties in California, USA. We classified these populations into two groups: restoration lakes with known fish stocking histories and recent experimental fish removal, and fishless lakes with a limited or absent fish stocking history (Sarnelle and Knapp 2004; Fisk et al. 2007). The study lakes are located in the Delaney Creek and Humphreys Basins at elevations ranging from 3150 to 3602 m and were chosen based on the results of previous zooplankton sampling (Stoddard 1987; Knapp et al. 2001b, 2005). Three of these lakes have no history of salmonid introductions: “Snowpole” [350430 E, 4123368 N (all coordinates are in UTM Zone 11)]; “Source” (349988 E, 4125708 N); and Middle Skelton (298120 E, 4201298 N). One lake, Wahoo Lake #2 (347968 E, 4121602 N), was stocked a single time in 1960 without follow-up stocking. This lake did not sustain a resident salmonid population following this initial introduction and is here considered a fishless population. The restoration lakes have a known history of salmonid introductions including golden trout (O. m. aguabonita) and brook trout (Salvelinus fontinalis). Marmot Lake was stocked with golden trout every other year from 1955 to 1995. Square Lake was stocked with brook trout in 1948 and with golden trout ever other year from 1955 to 1995. Fish were experimentally removed from Square Lake in 1997 and from Marmot Lake in 1998 [details of fish removal can be found in Sarnelle and Knapp (2004, 2005) and Knapp et al. (2007)].
Daphnia melanica were collected from the study lakes using a 30 cm conical tow net and either immediately preserved in 95% ethanol [Square and Marmot Lake 1999 samples (Sarnelle and Knapp 2004)], or transported to the laboratory where they were maintained at 4°C for a period of 1–2 weeks prior to isolation for establishment into clonal culture. All study lakes were sampled in 2005 and both restoration lakes were sampled in 1999, 1–2 years after fish removal and immediately after the first detection of D. melanica in zooplankton samples. To maximize the amount of genetic variation captured in live collections from each population, mature females from the original field collections were isolated and placed singly into 250 ml beakers containing 200 ml of filtered well-water. This procedure ensured that no isolates were genotypically identical individuals derived from asexually produced clutches released during the period from collection in the field until isolation in the lab. At 4°C, asexual offspring released prior to isolation in the lab would not have sufficient time to reach maturity and were discarded. Stock cultures were established and maintained from these isolated individuals by clonal reproduction under constant conditions of temperature (15°C) and photoperiod (16L:8D). D. melanica were fed a vitamin-supplemented, pure culture of the green alga Scenedesmus obliquus every 3–4 days.
Molecular methods and analysis
DNA extraction, PCR amplification, and sequencing: Genomic DNA from 20 individuals/population (24 individuals for Marmot 1999 and Square 1999; 168 individuals total) was obtained by a standard proteinase-K digestion followed by phenol/chloroform/isoamyl alcohol extraction (Sambrook and Russell 2002).
Microsatellite loci: The polymerase chain reaction (PCR) was used to amplify six tri-nucleotide microsatellite markers (Colbourne et al. 2004) in 24 individuals from the restoration populations collected in 1999 (Square Lake and Marmot Lake) and 20 individuals collected from all populations in 2005. Amplifications were performed in 25 μl reactions using 1 μl of genomic DNA extraction, 2.5 μl 10× PCR buffer with MgCl2, 2.5 μl 8 mM dNTP mix, 2.5 μl of each 2 μM primer, 0.1 μl Taq DNA polymerase (Perkin Elmer Cetus), and water to final volume. Reactions consisted of an initial denaturation step at 94°C for 4 min, followed by 36 cycles of 92°C for 60 s, locus-specific annealing temperature for 60 s, and 72°C for 60 s, and a final extension step at 72°C for 10 min. Negative controls were performed with all reactions. Primer sequences and locus-specific annealing temperatures are given in Table 1. Fluorescently labeled amplicons were run with a ROX 400 (Applied Biosystems Inc) size standard on an ABI 3100 automated sequencer. Individual genotype profiles were visualized and scored for each primer combination using GeneMapper v4.0 software (Applied Biosystems Inc).
Mitochondrial genes: The polymerase chain reaction (PCR) was used to amplify portions of the mtDNA COI and ND5 genes using primers COIf (5′-TCT CAA CTA CTC ATA AGG ACA TTG G-3′), COIr (5′-TAT ACT TGA GGA TGA CCA AAG AAC CA-3′), ND5a (5′-AAA GAA GAA ACC ATA TTA AAC C-3′), and ND5b (5′-GGG GTG TAT CTA TTA ATT CG-3′). Mitochondrial sequences were generated from ten individuals in all populations except Middle Skelton Lake where sequences were generated from nine individuals. Amplifications were performed in 50 μl reactions using 2 μl of genomic DNA extraction, 5 μl 10× PCR buffer with MgCl2, 5 μl 8 mM dNTP mix, 5 μl of each 2 μM primer, 0.2 μl Taq DNA polymerase (Perkin Elmer Cetus), and water to final volume. Reactions consisted of an initial denaturation step at 94°C for 5 min, followed by 35 cycles of 92°C for 45 s, 46°C for 60 s, and 72°C for 90 s, and a final extension step at 72°C for 5 min. Negative controls were performed with all reactions. PCR products were purified using Qiaquick PCR purification spin columns (QIAGEN Inc.). The DNA sequences of purified PCR products were generated using Dye Terminator Cycle Sequencing (Applied Biosystems Inc) and run on an ABI 3100 automated DNA sequencer (Applied Biosystems Inc). PCR products were sequenced from both the forward and reverse directions to generate overlapping fragments, which were assembled into contiguous sequences and aligned using SEQUENCHER 4.2.2 (Gene Codes). The final alignment of 1325 bp comprised 618 bp of COI and 707 bp of ND5. All unique mtDNA sequences were deposited in GenBank (accession HM 131366-131425, HM 137667-137726).
We evaluated the level of variability for microsatellite data by generating estimates of the expected heterozygosity (He), and gene diversity (π) in ARLEQUIN (Schneider et al. 2000). Population specific estimates of expected heterozygosity were calculated as the average over six loci. We also calculated population-specific estimates of the degree of homozygosity in excess of Hardy–Weinberg expectations (Gis) as a metric to describe the degree to which populations deviate from Hardy–Weinberg equilibrium. For estimates of Gis, positive values imply heterozygote excess while negative values suggest homozygote excess. Estimates of average gene diversity (π) over all loci for each population were calculated as the probability that two randomly chosen homologous loci are different (Nei 1987). Our estimates of gene diversity for microsatellite data are equivalent to estimates of gene diversity at the nucleotide level for sequence data. To ascertain the level of population differentiation we estimated Dest (Jost 2008, 2009) rather than the traditional measures of population subdivision (i.e. Gst and related measures) because Dest does not rely on the assumptions inherent in the traditional measures of population differentiation that are often violated, particularly in clonally reproducing Daphnia populations. Furthermore, a meta-analysis of 34 published studies showed that Dest is the most appropriate estimator of population differentiation for studies utilizing microsatellite markers (Heller and Siegismund 2009). We used SMOGD v1.2.5 (Crawford 2010) to calculate estimates of Dest. Finally, we evaluated the current level of genetic variability in mitochondrial sequence data from each of our populations by generating estimates of nucleotide diversity (πN; Nei 1987) in ARLEQUIN 2.0 (Schneider et al. 2000).
Common-garden experiment and quantitative-genetic analysis
Morphological (body size at maturity) and life-history (age at maturity and number of offspring in first clutch) traits were assayed in a subset of our study populations collected in 2005 using a standard experimental design (Lynch 1985; Pfrender and Lynch 2000). The subset of populations used included two restoration populations (Marmot and Square) and two fishless populations (Snowpole and Source). Single immature females were isolated from unique clonal stock cultures from each population. These individuals were maintained as single, asexually produced progeny for two generations. Morphological and life-history traits were assayed in third generation individuals. This design ensures maternal and grand-maternal effects will contribute to the within-clone rather than among-clone component of variance. Fifty clones randomly chosen from the stock cultures of each population were assayed in the common garden. Each clone had two replicates yielding 250 clonal lines and 500 total individuals.
Individuals were maintained in 250 ml beakers containing 150 ml filtered well-water supplemented with S. obliquus at a concentration of 97.5% light transmittance. The water-algae mixture was replaced every other day to ensure constant food density. Beakers were kept on randomly placed trays in a environmental chamber at 18°C and a photoperiod of 16L:8D. The position of trays was changed daily to minimize micro-environmental differences in the chamber.
Body size at maturity (first appearance of eggs in brood pouch), from the top of the head to the base of the tail spine, was measured with a stereomicroscope and an optic ruler calibrated using a micrometer slide. Upon release of the first clutch, the number of live offspring was counted. Age estimates were calculated by subtracting the date at maturity from the date of birth and refined by identifying the embryonic developmental stage (Spitze 1993; Lynch et al. 1999).
Population specific broad-sense heritabilities for each trait were estimated using one-way ANOVA for unbalanced design to partition the total phenotypic variance into the within-clone and among-clone components, respectively the environmental and genetic sources of variation. Broad-sense heritability was then estimated as H2 = Vg/(Vg + Ve). Standard errors for H2 were estimated by the delta method (Lynch and Walsh 1998).
Results
Molecular genetic variation
In general, estimates of genetic diversity based on microsatellite loci were lower in restoration populations than historically fishless populations (Table 2). A majority of populations had at least one locus that significantly deviated from Hardy–Weinberg expectations. Population group averages of homozygosity in excess of Hardy–Weinberg expectations (Gis) in restoration populations (1999—0.13; 2005—0.14) and fishless populations (0.15) were similar, both showing an excess of heterozygotes. Estimates of gene diversity (π) were low in restoration populations relative to fishless populations.
The level of population differentiation based on microsatellite markers among populations was similar for the dataset that included all fishless populations and only the restoration population samples from 1999 (Dest = 0.17) and the dataset that included all fishless populations and only the restoration populations sampled in 2005 (Dest = 0.18). However, Middle Skelton Lake is located in a different basin than the remaining lakes, and when data from this lake is removed so that only lakes located in the Humphreys Basin are included, estimates of differentiation decrease substantially (1999, Dest = 0.08; 2005, Dest = 0.10). Within each restoration lake across the sampling interval, the level of population differentiation for Marmot Lake from 1999 to 2005 was 0.01 while the level of population differentiation for Square Lake from 1999 to 2005 was 0.03.
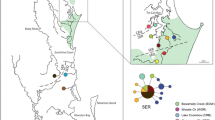
The concatenated COI and ND5 sequences revealed 19 unique mitochondrial haplotypes. Populations within Humphreys Basin (i.e. excluding Middle Skelton Lake) contained 16 unique mtDNA haplotypes. The restoration populations (Square and Marmot Lakes) both had a combination of unique and shared haplotypes (Fig. 1). Square Lake contained three different haplotypes, one unique and two shared with Snowpole. Marmot Lake had higher haplotype diversity with five different haplotypes. Three of these haplotypes were unique to Marmot Lake, one shared with Source, and one shared with Wahoo Lake. The fishless populations (Source Lake, Snowpole Lake, and Wahoo Lake) also showed a pattern of unique and shared haplotypes. Source Lake had two haplotypes, one unique and one shared with Marmot Lake. Snowpole Lake had eight haplotypes, six were unique, while the remaining two were shared with Square Lake. Wahoo Lake had two haplotypes, one unique and the other shared with Marmot Lake. Estimates of nucleotide diversity (πN) in restoration populations were lower than estimates from fishless populations (Table 2).

Map of study populations and results of haplotype sharing. A portion of the Humphreys Basin is shown with study lakes dark shaded. The proportion of unique (black) and shared (white) mtDNA haplotypes for each population located in the Humphreys Basin are depicted by the bar. Middle Skelton Lake is not in the Humphreys Basin and is not shown. Inset shows the location of the study lakes in California, USA
Quantitative genetic variation
Broad-sense heritability estimates averaged over all traits were similar for both population types (Table 3). Overall, when all traits and all populations of a specific population type (fishless or restoration) were pooled, there were no significant differences among population types (ANOVA: df = 1, F = 0.00, P = 0.97). In restoration populations, the average level of genetic variation for body size (0.31) and age at maturity (0.28) is higher than the average estimates obtained for fishless populations (body size = 0.29; age at maturity = 0.22). However, genetic variation for offspring production appears to accrue slowly as the average level of variation in restoration populations (0.06) is lower than the average in fishless populations (0.13).
Discussion
Introduced species can cause severe demographic reductions in native populations which result in a loss of genetic diversity. For a majority of species faced with a bottleneck or local extinction, the primary avenues of reconstituting genetic variation are mutation and migration. The first, mutation, operates on exceedingly slow time scales and is relatively unimportant when considering management strategies to rapidly replenish variation in genetically depauperate populations. Indeed, the regeneration of single locus morphological or allozyme diversity can take in excess of 105 generations (Lande and Barrowclough 1987). Alternatively, recolonization and migration can rapidly increase genetic diversity.
However, some species, specifically those that produce dormant resting eggs, appear resilient to a bottleneck or local extinction of the active population. Hatching from diapausing propagules can result in rapid demographic recovery once a selective threat is alleviated (Knapp et al. 2001b; Sarnelle and Knapp 2004; Mergeay et al. 2007). In this study we examined the genetic status of two D. melanica populations in the Sierra Nevada following the experimental removal of nonnative fish to determine whether the rapid demographic recovery of these populations, attributed to hatching from the local egg bank (Sarnelle and Knapp 2004), also involved the rapid reacquisition of genetic diversity. Our data suggests that restoration populations rapidly acquired neutral and quantitative genetic variation, and that the levels of variation we observed are qualitatively slightly lower than, or comparable to, surrounding populations without a history of fish introductions. Estimates of genetic diversity based on nuclear markers in restoration populations sampled immediately following fish removal and roughly 8 years following fish removal are slightly depressed compared to other local populations. Mitochondrial sequence diversity is, on average, lower in restoration populations than in fishless populations. However, there is a large amount of variation in population-specific estimates of mitochondrial sequence diversity within population groups, hence genetic diversity appears broadly similar between population types. Additionally, estimates of broad-sense heritability averaged over three ecologically important traits in restoration populations are not significantly different from fishless populations. Taken together, these data demonstrate that planktonic D. melanica populations extirpated by introduced fish undergo rapid demographic recovery and that concomitant with demographic recovery is the generation of high levels of nuclear, mitochondrial, and quantitative genetic diversity following the experimental removal of fish.
These observations highlight the rapidity of reacquisition of genetic variation possible in natural populations, and add to a small number of studies examining this process. Similar to our alpine lake populations, where the environmental factor directly contributing to planktonic extirpation was removed, populations of dogwhelk locally extirpated by tributyltin (TBT) pollution attained levels of genetic diversity similar to unexposed populations within 10 years following a ban on TBT (Colson and Hughes 2004). The build up of genetic variation over longer time spans has also been demonstrated. For example, two species of mantis shrimp that became locally extinct following the volcanic eruption on Krakatau in 1883 were able to recolonize and regenerate levels of genetic diversity similar to nearby undisturbed populations over a 100-year period (Barber et al. 2002). The short-term reconstitution of adequate standing quantitative genetic variation is particularly relevant for populations experiencing rapid or continually changing environments since the level of standing variation may be a key determinant of population persistence (Gomulkiewicz and Holt 1995).
In Daphnia there are two sources of immigrants to recolonize lakes in which a planktonic population has been extirpated. Immigration can occur through dispersal of diapausing eggs from neighboring populations, or via hatching of diapausing eggs produced by the historical resident population that reside in lake bottom sediments. Daphnia populations are often characterized by high levels of population subdivision with a low effective migration rate, even at distances of a few kilometers (e.g., Lynch et al. 1999). The low effective migration rate often observed among established populations may reflect limited dispersal ability or it may be the result of selective filtering and strong local adaptation in resident populations (De Meester et al. 2002; Michels et al. 2003). The influence of selective filtering and local adaptation to biotic and abiotic factors can be seen in the high levels of population subdivision observed in many lake dwelling species of Daphnia despite evidence for high avian-mediated dispersal (Taylor et al. 1998) and high rates of colonization in new habitats lacking a diapausing egg bank (Louette and De Meester 2005). A second mechanism of recolonization in Daphnia is hatching from the reservoir of diapausing eggs produced previously by the extirpated planktonic population. Local egg banks in Daphnia can lead to rapid demographic recovery of bottlenecked or extirpated planktonic populations (Sarnelle and Knapp 2004; Mergeay et al. 2007). Furthermore, hatching from egg banks contributes to the maintenance of high levels of quantitative genetic variation for adaptive traits (Pfrender and Lynch 2000).
Our results that show relatively low levels of population differentiation, particularly among populations located in the Humphreys Basin where three fishless populations and both restoration populations are located, stand in contrast to previous results obtained for other groups of Daphnia populations. These low levels of population differentiation could simply be an artifact of the use of different metrics, we use Dest while most other studies use Gst and its analogs, or the low level of differentiation could reflect an atypically high level of gene flow among populations. Furthermore, the general lack of unique haplotype diversity in the restoration populations (Square—33% unique; Marmot—60% unique) could also be attributed to gene flow among populations. No direct estimates of dispersal in this lake system are available so we cannot ascertain the relative effects of limited dispersal and filtering via strong local adaptation on the level of population differentiation. Therefore, it is not possible to unambiguously distinguish the source of colonizing genotypes given these data, and it appears likely that both migration and hatching from the local egg bank have jointly determined the levels of genetic diversity in the restoration lakes. Immigrants from neighboring populations would tend to homogenize the restoration populations relative to the surrounding populations, consistent with our results of low population differentiation and minimal unique mitochondrial diversity in the restoration populations, while hatching from the egg bank would tend to preserve locally-adapted genotypes.
The restoration populations involved in this study were previously the subject of a study on the patterns of demographic recovery (Sarnelle and Knapp 2004). Demographic recovery in these lakes was similar in that first detection of individuals in both lakes occurred within 1 year of fish removal, and after 4 years the densities of D. melanica were approximately 5,000 individuals/m3 in both lakes. Combined with the results of this study it is clear that following the experimental removal of fish, extirpated planktonic populations of D. melanica are able to rapidly recover demographically, and generate substantial standing genetic diversity. Both of the restoration populations showed a high level of genetic resilience and were equally efficient at rapidly reconstituting molecular and quantitative genetic variation. Despite the profound demographic perturbation imposed by the introduction of a novel predator into this ecosystem these populations were able to maintain substantial levels of genetic variation. Importantly, organisms with a diapausing egg (or seed) bank, and with the capacity for asexual reproduction, may be resilient to novel selective regimes and display a high potential to re-establish genetic variation lost during demographic collapse or local extirpation.
References
Barber PH, Moosa MK, Palumbi SR (2002) Rapid recovery of genetic diversity of stomatopod populations on Krakatau: temporal and spatial scales of marine larval dispersal. Proc R Soc Lond B 269:1591–1597
Bell G, Gonzalez A (2009) Evolutionary rescue can prevent extinction following environmental change. Ecol Lett 12:942–948
Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708
Bonnell ML, Selander RK (1974) Elephant seals: genetic variation and near extinction. Science 184:908–909
Bradford DF, Cooper SD, Jenkins TM, Kratz K, Sarnelle O, Brown AD (1998) Influences of natural acidity and introduced fish on faunal assemblages in California alpine lakes. Can J Fish Aquat Sci 55:2478–2491
Colbourne JK, Robison B, Bogart K, Lynch M (2004) Five hundred and twenty-eight microsatellite markers for ecological genomic investigations using Daphnia. Mol Ecol Notes 4:485–490
Colson I, Hughes RN (2004) Rapid recovery of genetic diversity of dogwhelk (Nucella lapillus L.) populations after local extinction and recolonization contradicts predictions from life-history characteristics. Mol Ecol 13:2223–2233
Cousyn C, De Meester L, Colbourne JK, Brendonck L, Verschuren D, Volckaert F (2001) Rapid, local adaptation of zooplankton behavior to changes in predation pressure in the absence of neutral genetic changes. Proc Natl Acad Sci USA 98:6256–6260
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
De Meester L, Gomez A, Okamura B, Schwenk K (2002) The monopolization hypothesis and the dispersal-gene flow paradox in aquatic organisms. Acta Oecol 23:121–135
Donald DB, Vinebrooke RD, Anderson RS, Syrginnis Graham MD (2001) Recovery of zooplankton assemblages in mountain lakes from the effects of introduced sport fish. Can J Fish Aquat Sci 58:1822–1830
Fisk D, Latta LC IV, Knapp RA, Pfrender ME (2007) Rapid evolution in response to introduced predators I: rates and patterns of morphological and life-history trait divergence. BMC Evol Biol 7:22
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, UK
Gomulkiewicz R, Holt RD (1995) When does evolution by natural selection prevent extinction? Evolution 49:201–207
Heller R, Siegismund HR (2009) Relationship between three measures of genetic differentiation Gst, Dest and G′st: how wrong have we been? Mol Ecol 18:2080–2083
Hendry AP, Kinnison MT (1999) The pace of modern life: measuring rates of contemporary microevolution. Evolution 53:1637–1653
Hoelzel AR, Halley J, O’Brien SJ et al (1993) Elephant seal genetic variation and the use of simulation models to investigate historical population genetics. J Hered 84:443–449
Hoffman RL, Larson GL, Samora B (2004) Responses of Ambystoma gracile to the removal of introduced nonnative fish from a mountain lake. J Herpetol 38:578–585
Jost L (2008) Gst and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jost L (2009) D vs. Gst: response to Heller and Siegismund (2009) and Ryman and Leimar (2009). Mol Ecol 18:2088–2091
Knapp RA, Matthews KR (2000) Non-native fish introductions and the decline of the mountain yellow-legged frog from within protected areas. Conserv Biol 14:428–438
Knapp RA, Sarnelle O (2008) Recovery after local extinction: factors affecting re-establishment of alpine lake zooplankton. Ecol Appl 18:1850–1859
Knapp RA, Corn PS, Schindler DE (2001a) The introduction of nonnative fish into wilderness lakes: good intentions, conflicting mandates, and unintended consequences. Ecosystems 4:275–278
Knapp RA, Matthews KR, Sarnelle O (2001b) Resistance and resilience of alpine lake fauna to fish introductions. Ecol Monogr 71:401–421
Knapp RA, Hawkins CP, Ladau J, McClory JG (2005) Fauna of Yosemite National Park lakes has low resistance but high resilience to fish introductions. Ecol Appl 15:835–847
Knapp RA, Boiano DM, Vredenburg VT (2007) Removal of nonnative fish results in population expansion of a declining amphibian (mountain yellow-legged frog, Rana muscosa). Biol Conserv 135:11–20
Kohn MH, Murphy WJ, Ostrander EA, Wayne RK (2006) Genomics and conservation genetics. TREE 21:629–637
Lande R, Barrowclough GF (1987) Effective population size, genetic variation, and their use in population management. In: Soule ME (ed) Viable populations for conservation. Cambridge University Press, UK, pp 87–123
Louette G, De Meester L (2005) High dispersal capacity of cladoceran zooplankton in newly created communities. Ecology 86:353–359
Lynch M (1985) Spontaneous mutations for life-history characters in an obligate parthenogen. Evolution 39:804–818
Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer Associates, Sunderland
Lynch M, Pfrender ME, Spitze K et al (1999) The quantitative and molecular genetic architecture of a subdivided species. Evolution 53:100–110
Mergeay J, Vanoverbeke J, Verschuren D, De Meester L (2007) Extinction, recolonization, and dispersal through time in a planktonic crustacean. Ecology 88:3032–3043
Michels E, Audenart E, Ortells R, De Meester L (2003) Population genetic structure of three pond-inhabiting Daphnia species on a regional scale (Flanders, Belgium). Freshw Biol 48:1825–1839
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Orr HA, Unckless RW (2008) Population extinction and the genetics of adaptation. Am Nat 172:160–169
Parker BR, Schindler DW, Wilhelm FM (1996) Recovery of Hesperodiaptomus arcticus populations from diapausing eggs following elimination by stocked salmonids. Can J Fish Aquat Sci 74:1292–1297
Parker BR, Schindler DW, Donald DB, Anderson RS (2001) The effects of stocking and removal of a nonnative salmonid on the plankton of an alpine lake. Ecosystems 4:334–345
Pfrender ME, Lynch M (2000) Quantitative genetic variation in Daphnia: temporal changes in genetic architecture. Evolution 54:1502–1509
Sakai AK, Allendorf FW, Holt JS (2001) The population biology of invasive species. Ann Rev Ecol Syst 32:305–332
Sambrook J, Russell DW (2002) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Sarnelle O, Knapp RA (2004) Zooplankton recovery after fish removal: limitations of the egg bank. Limnol Oceanogr 49:1382–1392
Sarnelle O, Knapp RA (2005) Nutrient recycling by fish versus zooplankton grazing as drivers of the trophic cascade in alpine lakes. Limnol Oceanogr 50:2032–2042
Schindler DW, Parker BR (2002) Biological pollutants: alien fishes in mountain lakes. Water Air Soil Pollut Focus 2:379–397
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis. Ver 3.01. Genetics and Biometry Lab, Department of Anthropology, University of Geneva, Geneva
Spitze K (1993) Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135:367–374
Stoddard JL (1987) Microcrustacean communities of high-elevation lakes in the Sierra Nevada, California. J Plankton Res 9:631–650
Tallmon DA, Luikart G, Waples RS (2004) The alluring simplicity and complex reality of genetic rescue. Trends Ecol Evol 19:489–496
Taylor DJ, Finston TL, Hebert PDN (1998) Biogeography of a widespread freshwater crustacean: pseudocongruence and cryptic endemism in the North American Daphnia laevis complex. Evolution 52:1648–1670
Thompson JN (1998) Rapid evolution as an ecological process. Trends Ecol Evol 13:329–332
Vredenburg VT (2004) Reversing introduced species effects: experimental removal of introduced fish leads to rapid recovery of a declining frog. Proc Natl Acad Sci USA 101:7646–7650
Vredenburg VT, Bingham R, Knapp R et al (2006) Concordant molecular and phenotypic data delineate new taxonomy and conservation priorities for the endangered mountain yellow-legged frog. J Zool 4:361–374
Weber D, Stewart B, Garza J, Lehman N (2000) An empirical genetic assessment of the severity of the northern elephant seal population bottleneck. Curr Biol 10:1287–1290
Acknowledgements
We thank M. Kanaga, K. Reed, J. Thompson, and R. Wilcox for providing laboratory assistance. J. Mergeay, two anonymous reviewers, and the Utah State University Evolution Group provided helpful comments on the manuscript. Funding for this study was provided by National Science Foundation grants DEB-021212487 to M. E. Pfrender, and DEB-0075509 to R. Knapp and O. Sarnell.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Latta, L.C., Fisk, D.L., Knapp, R.A. et al. Genetic resilience of Daphnia populations following experimental removal of introduced fish. Conserv Genet 11, 1737–1745 (2010). https://doi.org/10.1007/s10592-010-0067-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0067-y