
Abstract
Habitat fragmentation is believed to be a key threat to biodiversity as it decreases the probability of survival of populations, reduces gene flow among populations and increases the possibility of inbreeding and loss of genetic diversity within populations. Heathlands represent excellent systems to study fragmentation effects as the spatial and temporal course of fragmentation is well documented for these habitats. At the beginning of the nineteenth century, heathlands were widespread in northern Germany, but they became increasingly fragmented at the end of the nineteenth century until only few fragments had been left. As many insect species are strongly specialized on heathland habitats, they represent ideal study systems to test the genetic effects of such recent fragmentation processes. The solitary bee Andrena fuscipes is strongly specialized on heather (Calluna vulgaris) and, therefore, occurs exclusively in heathland habitats. The species is red-listed in Germany and other parts of Europe. Here, we present an analysis of the genetic structure of 12 populations of A. fuscipes using eight microsatellite loci. The populations showed little geographical structure and the degree of genetic differentiation was low. Compared to related bee species, inbreeding coefficients were relatively low and seem to be mainly affected by the bees’ solitary nesting behaviour.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat loss, degradation and fragmentation of habitats are major factors influencing the maintenance of biodiversity (Fahrig 2003; Tscharntke and Brandl 2004). Habitat fragmentation is believed to reduce gene flow among populations, leading to a loss of genetic diversity within populations and increased population differentiation (Fischer and Lindenmayer 2007). During the last century, the characteristic cultural landscape of Central Europe experienced a rapid transformation caused by socio-economic changes. Many semi-natural and traditional cultural habitats have been destroyed due to the intensification of agricultural land-use practices, including the use of fertilisers and new cultivation techniques (Bakker and Berendse 1999). Some traditional agricultural habitats, which had been shaped by centuries of human influence (e.g. extensive hay meadows, dry oligotrophic grasslands or heathlands), have meanwhile become rare and fragmented (Exeler et al. 2009). Species confined to such habitat types are, therefore, among those listed under the highest red list categories.
Heathlands have been created by human land use on oligotrophic sandy soils, including sheep grazing, burning and sod cutting. These open habitats reached their maximum distribution during the eighteenth and early nineteenth century and became particularly widespread in the Netherlands, parts of Great Britain and northern Germany (Fig. 1a). They rapidly declined at the end of the nineteenth century and meanwhile have become restricted to small, isolated patches (Webb 1989; Fig. 1b, c). At present, heathland habitats are listed as critically endangered in Germany and other parts of Europe (Riecken et al. 2006) and are protected by the EU Habitats Directive (EU directive 92/43/EEC). However, heathlands are still threatened by changes in land-use, but also by the increasing nutrient deposition and rapid succession by shrubs and trees (Sedláková and Chytrý 1999). The remaining heathland fragments often represent local hot spots of biodiversity, as many endangered plant and animal species are adapted to the ecological conditions of these open, xerothermic habitats (Usher 1992; Dupont and Nielsen 2006). The prevailing warm and dry conditions particularly support a species rich insect fauna. Nowadays, the persistence of species-rich heathlands is highly dependent on habitat management (Usher 1992; Hochkirch et al. 2007, 2008). Some large heathlands in northern Germany have only persisted due to a continuous management by sheep farming, burning or sod cutting, but the largest areas still exist because of a use as military training areas. In contrast, unmanaged sites are often dominated by old, degenerated stands of heather offering poor conditions for heathland specialists.

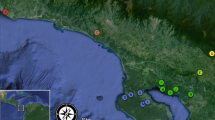
Decline of heathland habitats (grey) in north-western Germany and location of the study sites (black triangles). a Distribution of heathland at the beginning of the nineteenth century; b during the 1930s and c in 1980 (modified from Heckenroth 1985)
Given that heathland specialists are often unable to survive in other habitat types and the rapid fragmentation of their habitat is very well documented, populations of heathland species represent excellent model systems to study the genetic effects of habitat fragmentation. Wild bees are particularly interesting for such a study as a strong decrease in their abundance and species richness has been reported in many parts of the world (e.g. Biesmeijer et al. 2006, Cane and Tepedino 2001).This seems to be particularly true for species that are highly specialized on specific pollen sources as larval food or special habitat structures to build their nests (Exeler et al. 2009). As oligolectic bees are restricted to specific host plants, it has been proposed that they might be particularly prone to inbreeding and loss of genetic variability combined with a reduced gene flow among populations. The effects of habitat fragmentation on wild bee communities has often been studied (e.g. Steffan-Dewenter and Tscharntke 1999, Gathmann and Tscharntke 2002), but the consequences for the genetic population structure has not yet been investigated for heathland specialists. We studied the genetic effects of heathland fragmentation on Andrena fuscipes, a solitary bee which is specialized on heather pollen (Calluna vulgaris). Our main objective was to examine whether the relatively recent fragmentation influenced the genetic variability, the population genetic structure or the magnitude of gene flow among populations of this highly specialized species.
Methods
Study object
Andrena fuscipes is a solitary nesting bee species, which is univoltine with a flight period from August to September. In Central Europe, the species is specialized on heather pollen (Calluna vulgaris) as larval food. Females build single nests in the ground and prefer dry, sandy soils (Westrich 1989). Although A. fuscipes is widespread in Central Europe, it has become rare and is listed as vulnerable in countries where red lists for Hymenoptera exist, such as Germany (Westrich et al. 1998), the Netherlands (Peeters and Reemer 2003) and Ireland (Fitzpatrick et al. 2006).
Study area and sampling
Females of A. fuscipes were sampled on C. vulgaris during peak flight activity (between 11.00 a.m. and 5.00 p.m.) in August and September 2005 and 2006. Twelve locations in north-western Germany were sampled (Fig. 1), comprising a total of 195 individuals and spanning a geographical distance from less than 4 to 150 km (Table 1). We tried to collect at least 20 specimens in each population. However, A. fuscipes does not nest in high densities and some populations were too small to achieve this number. The study area is located in Lower Saxony and North Rhine-Westphalia. The sampling sites have been assigned to four regions. Five sites were located in the “Emsland”, four sites in the “Nordhorn” area and two in “Steinfurt”. In addition, we sampled one population at a greater distance (“Senne”). The whole study region is strongly characterized by intensive agricultural land use. The size and the type of management varied between the sampled locations. Two sites are used as military training areas, seven sites are extensively grazed by cattle or sheep and two sites are not managed at all.
DNA isolation and amplification
Genomic DNA was extracted from thoracic muscle tissue using the DNeasy Tissue Kit (Qiagen), following the manufacturers’ protocol. Each sample was genotyped at eight microsatellite loci (vaga01, vaga02, vaga08, vaga12, vaga13, vaga18, vaga20 and AJ01) developed by Paxton et al. (1996) and Mohra et al. (2000). The loci were amplified separately using the HotMasterMix (Eppendorf). The 5′-end of each forward primer set was labelled with a fluorescent marker, either 5-FAM, JOE or TAMRA. The products were genotyped on an ABI PRISM 377 automated DNA sequencer (Applied Biosystems, Inc.). Fragment lengths were determined using GENESCAN 3.1 and GENOTYPER 2.5 (Applied Biosystems, Inc.).
Statistical analysis
Tests for genotypic linkage disequilibrium were calculated with Fisher’s exact test and a Markov-chain method using GENEPOP 3.4 (Raymond and Rousset 1995). The presence of null alleles was inspected using MicroChecker 2.2.3 (Van Oosterhout et al. 2004). Allelic richness (R) and observed and expected heterozygosity (H O and H E respectively) for each population were calculated in GenAlEx 6.0 (Peakall and Smouse 2006), which performs a χ2 test to assess the significance of a departure from Hardy–Weinberg-Equilibrium (HWE). The measure of allelic richness R was used as it is independent of sample size (El Mousadik and Petit 1996). To evaluate inbreeding effects, the inbreeding coefficient (F IS) was calculated for each population within FSTAT 2.9.3 (Goudet 1995). To test whether the populations passed a recent genetic bottleneck we used the program Bottleneck 1.2.02 (Cornuet and Luikart 1996). A two-phased model of mutation (TPM) was applied in this analysis and the significance was determined by a Wilcoxon signed-rank test (1,000 replications). Since the size and the type of management (grazing, military, no management) varied between the sampled locations, we tested the effects of these traits on the allelic richness (R) and the inbreeding coefficient (F IS) with an analysis of covariance (ANCOVA) in the program “R 2.7.1” (R Development Core Team 2008).
The population genetic structure among the sampled populations was quantified with different methods. In a first step we quantified the magnitude of gene flow and the influence of geographical distance between each pair of population. Therefore the differentiation between pairs of populations was examined with a log-likelihood based exact test (G-test), which tests the distribution of genotypes between each pair of population as implemented in FSTAT (Goudet 1995). The significance of these tests was adjusted using standard Bonferroni corrections. This method seems to be particularly efficient for non-random mating populations and a low overall population structure (Petit et al. 2001). To test for isolation-by-distance, we calculated pairwise F ST distances based upon Wright’s F-statistics (Wright 1951) with the program GenAlEx 6.0 and tested these for a correlation with geographical distances. A Mantel-test of matrix correlations and a reduced major axis (RMA) regression to estimate the intercept and slope of the isolation-by-distance relationship was performed within the program IBD 1.52 (Bohonak 2002).
In a second step we were interested in the influence of the regional origin of populations on their genetic structure. Therefore Chord distances (DC) (Cavalli-Sforza and Edwards 1967) between each pair of population were calculated in Microsat 1.5b (Minch 1997) and visualized as an unrooted UPGMA tree in the program Phylip 3.57c (Felsenstein 1993) to check for a population clustering. Furthermore, we analysed the genetic structure among the sampled regions with a Bayesian model of population structure as implemented in BAPS 3.2 (Corander and Marttinen 2006). For this purpose we used an admixture based clustering method regarding the sampled regions (admixture based on pre-defined populations/regions). We defined three sampling regions, 1. Emsland (74 individuals), 2. Nordhorn (66 individuals) and 3. Steinfurt/Senne (55 individuals) and estimated the probability of the assignment of individuals to the corresponding cluster. BAPS uses a stochastic optimization algorithm which runs considerably faster than MCMC-based algorithms and has increased power to detect differentiation at small geographical distances (Corander and Marttinen 2006; Latch et al. 2006). Finally we evaluated these results by performing an analysis of molecular variance (AMOVA) in GenAlEx 6.0 (Peakall and Smouse 2006). The aim of the AMOVA was to perform a hierarchical analysis of variance to separate and test the levels of genetic diversity (diversity among groups of populations, among the populations within these groups and among the individuals within a population). Specifically, we estimated the genetic diversity among the sampled regions (Emsland, Nordhorn and Steinfurt together with Senne), among the populations within these regions and among the individuals within a population.
Results
Genetic diversity
A departure from Hardy–Weinberg expectations (excess of homozygotes) was found in 37.5% of the 96 locus-population combinations. A potential presence of null alleles was determined in 23 of the 96 cases, but all loci did amplify in all individuals (Table S1—supplementary material). Tests for linkage disequilibrium were not significant after Bonferroni correction. Overall microsatellite variability was high (2–17 alleles per locus and population). Allelic richness ranged from 4.00 to 5.81 (averaged over all loci) and mean expected heterozygosity (H E) ranged from 0.56 to 0.69 (Table 1). Positive inbreeding coefficients (F IS) were detected in all populations, ranging from 0.04 to 0.35. We detected no effects of fragment size or the management type (grazing, military, no management) on allelic richness or inbreeding coefficient (Table 2). The bottleneck analysis revealed a significant excess of heterozygosity for one population (A2; P = 0.039), indicating a recent bottleneck event.
Differentiation among populations
Genetic differentiation was generally low (Table 3) with the highest pairwise population differentiation in the Emsland area (average F ST = 0.054). We found a weak but non-significant isolation-by-distance effect among the populations (Mantel test, r = 0.103, P = 0.134, Fig. 2).

Relation between gene flow and geographic distance among sites (F ST vs. geographic distance; r 2 = 0.103 P = 0.13)
The arrangement of populations visualized in the UPGMA tree (Fig. 3) reflects their geographic origin to some extent. While populations located in the Emsland area were differentiated (Fig. 3), the Steinfurt populations C1 and C2 clustered together with populations from the “Nordhorn” region. Population D1 (Senne) branched off with the greatest genetic distance to all other populations.

Unrooted UPGMA tree based on Cavalli-Sforza and Edwards’ chord distance (D c) derived from allele frequencies at eight microsatellite loci. Branch length reflects the genetic distance between populations
Genetic admixture analysis using BAPS revealed different degrees of admixture for the presumed clusters (Fig. 4) with cluster 2 (Nordhorn) showing the highest rate of admixture supporting a high genetic exchange. In cluster 1 and 3, 42% of the individuals were assigned to the corresponding region with a probability of more than 0.75. The AMOVA revealed that 94% of the variance was explained by variation within populations, 6% among populations within regions. No variance was explained by the regional arrangement of populations.

Assignment probabilities of individual genotypes from 12 populations to predefined clusters using BAPS
Discussion
Habitat fragmentation and genetic differentiation
Our analyses revealed only weak genetic differentiation among the populations of A. fuscipes, indicating that the relatively recent fragmentation of heathland habitats has reduced gene flow only marginally. The Emsland samples were stronger differentiated than populations in the Nordhorn or Steinfurt area despite the greater geographical distance between some of the latter populations. These results indicate that geographical distance per se could not explain the degree of genetic differentiation. In fact, we only found a weak (and non-significant) isolation-per-distance. Similar results have been found in other bee species, such as the bumblebees Bombus pascuorum and B. sylvarum and the sand bee Andrena vaga, where differences in the landscape structure accounted for a restricted gene flow rather than geographical distance (Widmer and Schmid-Hempel 1999; Ellis et al. 2006; Exeler et al. 2008). It is likely that the mobility of a species strongly influences the genetic structure among populations. Strong gene flow and genetic admixture across populations is often found among strong flyers, such as bees (Chapman et al. 2003), while populations of flightless insects often exhibit a clear geographic structure (Witzenberger and Hochkirch 2008; Hochkirch and Damerau 2009). A strong degree of admixture might be particularly true for solitary nesting bee species, which need to be more mobile in order to find mates. In addition, small populations of A. fuscipes might persist at forest edges or other places with local occurrence of C. vulgaris. Such sites might serve as stepping stones increasing the dispersal among populations.
A high number of studies on the effects of landscape structure and fragmentation on population differentiation have been carried out during the last decades, many of which revealed conflicting results (Keyghobadi et al. 2005). The degree of specialization is often assumed to have negative effects on dispersal ability and, consequently, the effects of fragmentation are believed to be stronger for habitat specialists than for generalists (e.g. Kitahara and Fujii 1994; Kelley et al. 2000). This has been particularly proposed for specialized wild bee species which are often specialized on single plant species or genera as larval food (Packer et al. 2005; Zayed et al. 2005). On the other hand, one might argue that specialized species may be adapted to the sparse occurrence of their resources and may avoid negative effects of a patchy habitat structure by a higher dispersal ability or a higher effective populations size (Peterson and Denno 1998; Sallé et al. 2007; Exeler et al. 2008). It is likely that heathland habitats were strongly fragmented before they were spread by human activities. Species adapted to such naturally fragmented habitats might have stronger dispersal abilities than those occurring naturally in strongly interconnected habitat types (e.g. forests). Interestingly, the genetic differentiation of A. fuscipes populations was even lower than in a related bee species, Andrena vaga, which is adapted to highly dynamic floodplain habitats (Exeler et al. 2008). Both species are specialized on a single pollen resource (Calluna and Salix, respectively), showing that a high degree of specialization is a poor predictor of genetic divergence between populations.
Due to the loss of heathland habitats, A. fuscipes has become threatened in parts of Europe. However, the extinction of populations of this bee is not only caused by the decrease of its floral host. A recent study of the bee communities in the Emsland has shown that A. fuscipes is often absent even if its host, C. vulgaris, is present (von der Heide and Metscher 2003). This illustrates that other factors than host plant occurrence are important predictors of the occurrence of this species, such as the availability of nesting sites or the size of the habitat, which must sustain a minimum viable population (Tscharntke 1992). Indeed, many heathland patches in northern Germany are small and dominated by old, degenerated and even-aged stands of heather. The dense vegetation reduces the availability of suitable nesting sites. It has also been shown in other insect species that vegetation structure is often a more important component of the habitat than vegetation composition (e.g. Gröning et al. 2007). Thus, the suitability of heathlands for sustaining a diverse insect fauna is particularly influenced by habitat management (Usher 1992; Assmann and Janssen 1999, Hochkirch et al. 2008). Sheep grazing, burning and sod cutting are important management types in heathland habitats as they create a high structural heterogeneity. Non-managed heathlands may lose many insect species as the heather stands become dense and high, shading open sand patches, which are particularly important for insects adapted to xerothermic conditions (Wuellner 1999). Although we did not find any effects of management types on the genetic diversity of A. fuscipes populations, local extinction processes of this species might be affected by the reduction of potential nesting sites or decreasing nectar and pollen availability due to the aging of heather plants (von der Heide and Metscher 2003).
Genetic variability and inbreeding
Compared to microsatellite analyses in other bee species, A. fuscipes populations had a similar or even higher genetic variability (Paxton et al. 1996; Beveridge and Simmons 2006; Exeler et al. 2008). These results indicate that the populations of A. fuscipes are either still large enough to maintain a relatively high genetic diversity or that the high variability is a consequence of the former widespread distribution of Calluna heathlands in northern Germany (Fig. 1), which might have supported large populations of A. fuscipes for a long time. The first hypothesis is supported by Peeters (pers. comm.), who found A. fuscipes still rather common in the Netherlands, which is adjacent to our study sites. We also found only little evidence for genetic bottlenecks. This possibility was suggested only for one small population in the Emsland (population A2), which occurred in a small heathland fragment with old stands of heather plants. It is possible that the relatively recent habitat fragmentation might reduce the chance to detect bottlenecks (Ellis et al. 2006). However, these values might also be affected by the small sample size due to the rare occurrence of the species.
In all populations, we detected significant reduced heterozygosities compared to HWE and positive inbreeding coefficients (ranging from 0.04 to 0.35). Nevertheless, deviations from HWE were rather sparse compared to other Hymenoptera (Paxton et al. 1996; Danforth et al. 2003; Zayed et al. 2005; Stow et al. 2007, Exeler et al. 2008). A reduced heterozygosity might be caused by several factors, including non-random mating, population subdivision and the presence of null alleles (Callen et al. 1993). Based upon our results, a population subdivision can be excluded, as little structure was detected in our analyses. Although the program MicroChecker suggested the occurrence of some null alleles in our data, all loci amplified in all individuals (i.e. there were no homozygous null alleles). Null alleles are a common problem in microsatellite analyses (Dakin and Avise 2004), but it has been shown that a low number of null alleles only slightly influences population genetic analyses (see also Stahlhut and Cowan 2004 and Carlsson 2008 for discussion). It seems to be more likely, that the excess of homozygotes is influenced by high levels of inbreeding, which has been reported for several bee species as a result of their nesting strategies (Paxton et al. 1996; Danforth et al. 2003). Compared to other bee species, the inbreeding coefficients of A. fuscipes were even relatively low. This might be the consequence of the solitary nesting behaviour of A. fuscipes. Males of solitary nesting bee species patrol at flowers to search for females, whereas males of species nesting in aggregations often stay at the nesting site to mate with hatching females (Exeler et al. 2008). Communally breeding species even stay in the natal nest to mate (Paxton 2005). As Hymenoptera are haplodiploid organisms, inbreeding might also be higher due to the reduced effective population sizes (Packer and Owen 2001).
Conclusions
Studies on fragmentation effects on the genetic structure and diversity of populations are common, but heathlands have rarely been studied in this context, although they are highly suitable for such analyses. Due to the strong losses of heathland habitats in northern Germany, we expected a high degree of genetic differentiation and a low degree of gene flow among the remaining heathland fragments. However, our results show that strong flyers, such as bees, might be only little affected by recent fragmentation events of former continuous habitats. The variable genetic differentiation observed among A. fuscipes populations might reflect different stages of an ongoing process of genetic isolation. The relatively high levels of genetic variability and relatively low levels of inbreeding might be affected by its solitary nesting behaviour.
References
Assmann T, Janssen J (1999) The effects of habitat changes on the endangered ground beetle Carabus nitens (Coleoptera: Carabidae). J Insect Conserv 3:107–116
Bakker JP, Berendse F (1999) Constraints in the restoration of ecological diversity in grassland and heathland communities. Trends Ecol Evol 14:63–68
Beveridge M, Simmons LW (2006) Panmixia: an example from Dawson’s burrowing bee (Amegilla dawsoni) (Hymenoptera: Anthophorini). Mol Ecol 15:951–957
Biesmeijer JC, Roberts SPM, Reemer M, Ohlemuller R, Edwards M, Peeters T, Schaffers AP, Potts SG, Kleukers R, Thomas CD, Settele J, Kunin WE (2006) Parallel declines in pollinators and insect-pollinated plants in Britain and the Netherlands. Science 313:351–354
Bohonak AJ (2002) IBD (Isolation by Distance): a program for analysis of isolation by distance. J Hered 93:153–154
Callen DF, Thompson AD, Shen Y, Phillips HA, Richards RI, Mulley JC, Sutherland GR (1993) Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am J Hum Genet 52:922–927
Cane JH, Tepedino VJ (2001) Causes and extent of declines among native North American invertebrate pollinators: detection, evidence, and consequences. Conserv Ecol 5:1
Carlsson J (2008) Effects of microsatellite null alleles on assignment testing. J Hered 99:616–623
Cavalli-Sforza LL, Edwards AWF (1967) Phylogenetic analysis. Models and estimation procedures. Am J Hum Genet 19:233–257
Chapman RE, Wang J, Bourke AFG (2003) Genetic analysis of spatial foraging patterns and resource sharing in bumble bee pollinators. Mol Ecol 12:2801–2808
Corander J, Marttinen P (2006) Bayesian identification of admixture events using multi-locus molecular markers. Mol Ecol 15:2833–2843
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for inferring recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Dakin EE, Avise JC (2004) Microsatellite null alleles in parentage analysis. Heredity 93:504–509
Danforth BN, Shuqing J, Ballard LJ (2003) Gene flow and population structure in an oligolectic desert bee, Macrotera (Macroteropsis) portalis (Hymenoptera: Andrenidae). J Kans Entomol Soc 76:221–235
Dupont YL, Nielsen BO (2006) Species composition, feeding specificity and larval trophic level of flower-visiting insects in fragmented versus continuous heathlands in Denmark. Biol Conserv 131:475–485
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Ellis JS, Knight ME, Darvill B, Goulson D (2006) Extremely low effective population sizes, genetic structuring and reduced genetic diversity in a threatened bumblebee species, Bombus sylvarum (Hymenoptera: Apidae). Mol Ecol 15:4375–4386
Exeler N, Kratochwil A, Hochkirch A (2008) Strong genetic exchange among populations of a specialist bee, Andrena vaga (Hymenoptera: Andrenidae). Conserv Genet 9:1233–1241
Exeler N, Kratochwil A, Hochkirch A (2009) Restoration of riverine inland sand dunes: implications for the conservation of wild bees (Apoidea). J Appl Ecol 46:1097–1105
Fahrig L (2003) Effects of habitat fragmentation on biodiversity. Annu Rev Ecol Syst 34:487–515
Felsenstein J (1993) Phylip (Phylogeny Inference Package) v3.57c. Department of Genetics, University of Washington, Seattle
Fischer J, Lindenmayer DB (2007) Landscape modification and habitat fragmentation: a synthesis. Glob Ecol Biogeogr 16:265–280
Fitzpatrick U, Murray TE, Byrne A, Paxton RJ, Brown MJF (2006) Regional red list of Irish bees. Report to National Parks and Wildlife Service (Ireland) and Environment and Heritage Service (N. Ireland). http://www.npws.ie/en/media/Media,4860,en.pdf. Accessed Nov 2006
Gathmann A, Tscharntke T (2002) Foraging ranges of solitary bees. J Anim Ecol 45:742–752
Goudet J (1995) FSTAT (Version 1.2): a computer program to calculate F statistics. J Hered 86:485–486
Gröning J, Krause S, Hochkirch A (2007) Habitat preferences of an endangered insect species, Cepero’s ground-hopper (Tetrix ceperoi). Ecol Res 22:767–773
Heckenroth H (1985) Atlas der Brutvögel Niedersachsens 1980. Naturschutz Landschaftspfl Niedersachs 14:1–428
Hochkirch A, Damerau M (2009) Rapid range expansion of a wing-dimorphic bush-cricket after the 2003 climatic anomaly. Biol J Linn Soc 97:118–127
Hochkirch A, Witzenberger K, Teerling A, Niemeyer F (2007) Translocation of an endangered insect species, the field cricket (Gryllus campestris Linnaeus, 1758) in northern Germany. Biodivers Conserv 16:3597–3607
Hochkirch A, Gärtner A-C, Brandt T (2008) Effects of management of degraded inland dunes on the endangered heath grasshopper, Chorthippus vagans (Orthoptera: Acrididae). Bull Entomol Res 98:449–456
Kelley ST, Farrell BD, Mitton JB (2000) Effects of specialization on genetic differentiation in sister species of bark beetles. J Hered 84:218–227
Keyghobadi N, Roland J, Strobeck C (2005) Genetic differentiation and gene flow among populations of the alpine butterfly, Parnassius smintheus, vary with landscape connectivity. Mol Ecol 14:1897–1909
Kitahara M, Fujii K (1994) Biodiversity and community structure of temperate butterfly species within a gradient of human disturbance: an analysis based on the concept of generalist vs. specialist strategies. Res Popul Ecol 36:187–199
Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE (2006) Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet 7:295–302
Minch E (1997) Microsat, version 1.5b. Stanford University Medical Centre, Stanford, CA
Mohra C, Fellendorf M, Segelbacher G, Paxton RJ (2000) Dinucleotide microsatellite loci for Andrena vaga and other andrenid bees from non-enriched and CT-enriched libraries. Mol Ecol 9:2189–2191
Packer L, Owen R (2001) Population genetic aspects of pollinator decline. Conserv Ecol 5. http://www.consecol.org/vol5/iss1/art4/
Packer L, Zayed A, Grixti JC, Ruz L, Owen RE, Vivallo F, Toro H (2005) Conservation genetics of potentially endangered mutualisms: reduced levels of genetic variation in specialist versus generalist bees. Conserv Biol 19:195–202
Paxton RJ (2005) Male mating behaviour and mating systems of bees: an overview. Apidologie 36:145–156
Paxton RJ, Thorén PA, Tengö J, Estoup A, Pamilo P (1996) Mating structure and nestmate relatedness in a communal bee, Andrena jacobi (Hymenoptera, Andrenidae), using microsatellites. Mol Ecol 5:511–519
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peeters TMJ, Reemer M (2003) Bedreigde en verdwenen bijen in Nederland (Apidae). European Invertebrate Survey, Nederland, Leiden
Peterson MA, Denno R (1998) The influence of dispersal and diet breadth on patterns of genetic isolation by distance in phytophagous insects. Am Nat 152:428–446
Petit E, Balloux F, Goudet J (2001) Sex biased dispersal in a migratory bat: a characterization using sex-specific demographic parameters. Evolution 55:635–640
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2008) R: a language and environment for statistical computing. http://www.R-project.org
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Riecken U, Finck P, Raths U, Schröder E, Ssymank A (2006) Rote Liste der gefährdeten Biotoptypen Deutschlands. Zweite fortgeschriebene Fassung 2006. Schriftenreihe Naturschutz und Biologische Vielfalt, vol 34. Bundesamt für Naturschutz, Bonn
Sallé A, Arthofer W, Lieutier F, Stauffer C, Kerdelhué C (2007) Phylogeography of a host-specific insect: genetic structure of Ips typographus in Europe does not reflect past fragmentation of its host. Biol J Linn Soc 90:239–246
Sedláková I, Chytrý M (1999) Regeneration patterns in a Central European dry heathland: effects of burning, sod-cutting and cutting. Plant Ecol 143:77–87
Stahlhut JK, Cowan DP (2004) Inbreeding in a natural population of Euodynerus foraminatus (Hymenoptera: Vespidae), a solitary wasp with single-locus complementary sex determination. Mol Ecol 13:631–638
Steffan-Dewenter I, Tscharntke T (1999) Effects of habitat isolation on pollinator communities and seed set. Oecologia 121:432–440
Stow A, Silberbauer L, Beattie AJ, Briscoe DA (2007) Fine-scale genetic structure and fire-created habitat patchiness in the Australian allodapine bee, Exoneura nigrescens (Hymenoptera: Apidae). J Hered 98:60–66
Tscharntke T (1992) Fragmentation of Phragmites habitats, minimum viable population size, habitat suitability, and local extinction of moths, midges, flies, aphids, and birds. Conserv Biol 6:530–536
Tscharntke T, Brandl R (2004) Plant–insect interactions in fragmented landscapes. Annu Rev Entomol 49:405–430
Usher MB (1992) Management and diversity of arthropods in Calluna heathland. Biodivers Conserv 1:63–79
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting errors in microsatellite data. Mol Ecol Notes 4:535–538
von der Heide A, Metscher H (2003) Zur Bienen- und Wespenbesiedlung von Taldünen der Ems und anderen Trockenstandorten im Emsland (Hymenoptera; Aculeata). Drosera 1:95–130
Webb NR (1989) Studies on the invertebrate fauna of fragmented heathland in Dorset, UK, and the implications for conservation. Biol Conserv 47:153–165
Westrich P (1989) Die Wildbienen Baden Württembergs. Eugen Ulmer, Stuttgart
Westrich P, Schwenninger HR, Dathe H, Riemann H, Saure C, Voith J, Weber K (1998) Rote Liste der Bienen (Hymenoptera, Apidae). In: Riecken U, Finck P, Raths U, Schröder E, Ssymank A (eds) Rote Liste gefährdeter Tiere Deutschlands, vol 55. Bundesamt für Naturschutz (Hrsg), Schriftenreihe Landschaftspflege und Naturschutz, pp 119–129
Widmer A, Schmid-Hempel P (1999) The population genetic structure of a large temperate pollinator species, Bombus pascuorum (Scopoli) (Hymenoptera: Apidae). Mol Ecol 8:387–398
Witzenberger KA, Hochkirch A (2008) Genetic consequences of animal translocations: a case study using the field cricket, Gryllus campestris L. Biol Conserv 141:3059–3068
Wright S (1951) The genetic structure of populations. Ann Eugen 15:323–354
Wuellner CT (1999) Nest site preference and success in a gregarious, ground nesting bee Dieunomia triangulifera. Ecol Entomol 24:471–479
Zayed A, Packer L, Grixti JC, Ruz L, Owen RE, Toro H (2005) Increased genetic differentiation in a specialist versus a generalist bee: implications for conservation. Conserv Genet 6:1017–1026
Acknowledgements
We are grateful to A. Schanowski, C. Venne and S. Hameister for collecting material of A. fuscipes. We thank U. Coja for help in the laboratory. K. A. Witzenberger and T. Eggers gave helpful comments on a previous version of this manuscript. We also wish to thank R. Paxton for providing microsatellite primers and helpful comments. Financial support was provided by the German Federal Environmental Foundation (“Deutsche Bundesstiftung Umwelt”) and the Environmental Foundation of Weser-Ems (“Umweltstiftung Weser-Ems”). We thank the regional administrations for the permission to collect specimens of A. fuscipes.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Exeler, N., Kratochwil, A. & Hochkirch, A. Does recent habitat fragmentation affect the population genetics of a heathland specialist, Andrena fuscipes (Hymenoptera: Andrenidae)?. Conserv Genet 11, 1679–1687 (2010). https://doi.org/10.1007/s10592-010-0060-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0060-5