
Abstract
The Norwegian red deer population (Cervus elaphus) was from the mid eighteenth to the early twentieth century drastically reduced in size and distribution but has the last century expanded both demographically and spatially. We have investigated genetic variation, differentiation and admixture in this spatially expanding ungulate population, using 14 microsatellites. The present genetic structure is moderate to strong with an average F ST = 0.08. Low M-ratios indicate loss of genetic variation in all localities and signals of a recent bottleneck was identified in 14 of 15 localities. Genetic distances between the localities indicate two main routes of dispersal during expansion, from the north–west and south–west, respectively. Bayesian assignment tests verify a break of the dataset in two, and demonstrate 99.9% probability for the existence of five sub-populations, which coincide well with five relict populations described by historic records. Computer simulations suggest that the observed genetic differentiation is recent rather than ancient, and that it may be explained by models of fragmentation or of founder events and subsequent merging rather than by models of recent bottlenecks in some particular demes within an ancient genetic structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The demography and distribution of species vary through time and space (Begon et al. 1996) and greatly affect levels of genetic variation and population structure (Hartl and Clark 1997; Hedrick 2000). Many species have a history of reduced or fragmented population size, often followed by demographic growth and spatial expansion. During the pleistocene, extensive climatic oscillations and rapid changes in the distribution of continental ice sheets resulted in successive shifts in the demography and geographical range of many species. Founder events and isolation after successive leading edge expansions involved loss of genetic variation and increased homozygosity in many of the newly colonised areas (Hewitt 2000, 2001). Recently, scientists have established significant climatic changes since pre-industrial times that also have involved population fluctuations and range shifts for many species, especially in temperate areas (IPCC 2001, 2007).
Loss of genetic variation during bottlenecks (Nei et al. 1975; Chakraborty and Nei 1977) may together with demographic factors in very small and fragmented populations involve reduced adaptability and increased risk of extinction (Lande 1988; Soulé and Mills 1992). The genetic effects of demographic population expansions have been well examined (Slatkin and Hudson 1991; Beaumont 1999; Chakraborty and Kimmel 1999), but recently attention has been drawn to the effects of spatial population expansion on genetic structure (Ray et al. 2003; Excoffier 2004). With a limited number of dispersing individuals genetic variation may be lost during colonisation because of founder effects and subsequent bottlenecks (Hedrick 2000). In expanding populations new demes may become genetically differentiated because of genetic drift depending on the migration rates (Austerlitz et al. 1997; Excoffier 2004), especially when dispersers move long distances and become isolated (Nichols and Hewitt 1994; Ibrahim et al. 1996). However, the homogenising effect of migration on genetic structure is large and when genetically different subpopulations merge, the level of genetic variation can increase as a result of the isolate break (Hartl and Clark 1997). Thus, under a spatial range expansion, genetic variation may be lost from founder effects and subsequent bottlenecks, but may also increase due to merging of genetically different demes. Genetic structure is one of the parameters for estimation of effective population size and is thus important for making management and conservation plans (Wang and Caballero 1999; Nunney 2000).
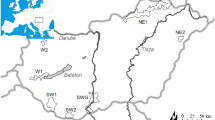
The red deer (Cervus elaphus) is an ungulate species and a highly priced game and trophy for hunting. In Norway the red deer (Cervus elaphus atlanticus) has existed at least since the Subboreal period (5,700–2,600; Collett 1909; Ahlèn 1965) and written records document an abundant population distributed throughout most of Southern Norway until approximately year 1750 (Friis 1874; Collett 1877). In the mid eighteenth century the Norwegian red deer population declined drastically and until the beginning of the last century it was confined to only five or six locations along the western coast (Fig. 1) counting a few hundred individuals in total at the most extreme (Collett 1909; Ingebrigtsen 1924). In southernmost Sweden a separate red deer population was reduced even more (Lønnberg 1906) and has for the last 150 years been confined to a very small population (Ahlèn 1965). Since the beginning of last century, and especially after 1950, the Norwegian red deer population has expanded from the western coast localities, demographically as well as spatially. It is now common in most parts of southern and central Norway with a total population size ranging from 100,000 to 120,000 individuals in 1997 (Langvatn 1988, 1998; Forchhammer et al. 1998; Fig. 1). Many reasons have been suggested for these population fluctuations, including high pressures of predation and hunting from the middle of the eighteenth century (Collett 1877, 1909), as well as temporal changes in the use of agricultural land (Ahlèn 1965; Mysterud et al. 2002). Here, we assess the present genetic variation of the Norwegian red deer population to look for any signatures of recent bottlenecks and to address the effect of spatial population expansion on genetic structure. We test the null hypothesis that demographic and spatial expansion has homogenised the population and that no genetic structure will be observed in spite of the previous population reduction. Rejection involves at least three alternative hypotheses for genetic structure (1) the recent fragmentation described by historic records, (2) bottlenecks in areas thought to be deprived of red deer and (3) founding events during spatial expansion.

Methods and materials
Sampling and laboratory procedures
Between 2000 and 2004 we sampled blood or tissue from 419 wild Norwegian red deer from 24 municipalities across Norway (Fig. 1). Samples from some of the municipalities were pooled to obtain a minimum of 15 individuals in each of totally 15 localities (Table 1). In general, the western localities are distributed within the area where the Norwegian red deer population was confined from the mid eighteenth to the early twentieth century, whereas the eastern localities are recently established populations outside this area (Table 1; Fig. 1).
Genomic DNA was isolated from whole blood and muscle tissue (Qiagen, DNeasy Kit). Previous investigations have indicated a generally low level of genetic variation in Norwegian red deer (Baccus et al. 1983; Gyllensten et al. 1983; Røed 1998; Røed and Midthjell 1998). We selected 14 polymorphic microsatellite loci that show Mendelian heredity in Norwegian red deer (Haanes et al. 2005). These were CSSM03 (Moore et al. 1994), OarCP26 (Ede et al. 1995), RT5 (Wilson et al. 1997), SRCRSP10 (Bhebhe et al. 1994), NVHRT73 and NVHRT48 (Røed and Midthjell 1998), McM58 (Hulme et al. 1994), OarFCB193 and OarFCB304 (Buchanan and Crawford 1993), BM5004, BM888, BMC1009, BM4208 and BM4107 (Bishop et al. 1994). The microsatellites were amplified on a GeneAmp PCR System 9600 (Applied Biosystems) in 10 μl reaction mixtures with 30–60 ng of genomic template DNA, 2 pmol of each primer, 50 mM KCl, 1.5 mM MgCl2, 10 mM Tris–HCl, 0.2 mM dNTP, and 0.5 U of AmpliTaq DNA polymerase (Applied Biosystems). Thermocycling parameters after denaturation at 94°C for 5 min were 30 cycles with 95°C for 1 min, 55°C for 30 s and 72°C for 1 min, followed by an additional 10 min at 72°C. The PCR products were then separated by size with capillary electrophoresis (ABI310, Applied Biosystems) and electromorphs were genotyped with GENOTYPER1.1.1 (Applied Biosystems).
Population genetics analysis
Each of the 15 localities was assessed through exact tests of Hardy–Weinberg equilibrium across the 14 loci using GENEPOP 3.4 with the default settings (Raymond and Rousset 1995). Sequential Bonferroni correction was used to adjust for repeated tests (Rice 1989). To assess differences in genetic variation among localities we used FSTAT 2.9.3 (Goudet 2001) to calculate the allelic richness (El Mousadik and Petit 1996) and the gene diversity (Nei 1987) for each locality across loci. To assess possible impact on genetic variation, recent bottlenecks were addressed using a one-tailed Wilcoxon test (10,000 iterations) as implemented in the software BOTTLENECK (Cornuet and Luikart 1996), which tests if the observed gene diversity is higher than expected at mutation-drift equilibrium from the number of observed alleles in each locality across loci. Most microsatellites fit a two-phase model of mutation (TPM) better than a strict stepwise mutation model (Di Rienzo et al. 1994) and we therefore used a TPM model with the default settings of 30% variation from the infinite allele model (IAM) and 70% from the stepwise-mutation-model (SMM). Secondly, the M-ratio (Garza and Williamson 2001) was calculated for each locality as the ratio between the observed number of alleles and the number of repeats in the allele size range of each locus, averaged across loci. This would give an indication of any loss of alleles during any recent population reductions.
F-statistics (e.g., Weir 1996) as implemented in FSTAT, with Bonferroni adjusted significance tests, were used to assess genetic structure within (FIS) and among (FST) localities. Pairwise geographical distances among localities (km) were calculated between longitude and latitude (http://jan.ucc.nau.edu/~cvm/latlongdist.html), and isolation-by-distance was assessed in GENEPOP by testing the correlation between geographical distances and pairwise FST/(1−FST) values. We used a Mantel test (Mantel 1967) in the implemented program ISOLDE (10,000 permutations) to test for significance. To further assess genetic differentiation we used the genetic distance DA (Nei et al. 1983), which is based on the geometric distances of populations on a multidimensional hypersphere independently of any mutation models (Nei 1987; Nei 2000). Distances (DA) were calculated between each pair of localities and a neighbour joining (NJ) tree built with 1,000 bootstrap replicates across loci with the software POPULATIONS (available at http://www.pge.cnrs-gif.fr/bioinfo/populations/index.php). The tree was visualised by the software TREEVIEW (Page 1996).
To assess genetic structure without prior knowledge of sampling locations, we used Bayesian assignment as implemented in STRUCTURE (Pritchard et al. 2000). The log likelihood of our data set (ln Pr(X∣K)) was estimated given different numbers of genetic clusters (K∈[1,7] using an admixture model (α = 1, αmax = 50) with uniform priors, in combination with either independent or correlated allele frequencies (Falush et al. 2003), using 100,000 burnin cycles and 500,000 MCMC iterations. For each K-value, STRUCTURE estimates the mean log likelihood of the data set (ln Pr(D∣K)) from several runs and uses Bayes’ theorem to compute the posterior probability of each K-value. Since higher K values often involve runs with higher posterior probabilities but a higher variance among runs (Evanno et al. 2005), we also identified K from the marked increase of variance among runs and calculated delta K to identify breakpoints in the data set. Genetic structure and the degree of admixture among the 15 localities were then interpreted from their membership in each of the K clusters and from the probabilities of individual assignment to these clusters using 0.5 as a threshold for assignment.
To address the hypotheses of present genetic structure we used the coalescent-based program SIMCOAL2.1.2 (Laval and Excoffier 2004) to simulate microsatellite diversity under three demographic scenarios. (1) A recent fragmentation into five demes. (2) Recent bottlenecks in two of five ancient demes. (2) A recent split into three demes that more recently separately found four smaller demes that merge pairwise through high migration (r = 0.5). In all three scenarios the initial population size is reduced 50 generations ago, and recent events involve low effective sizes for 30 generations before more recently growing to a size comparable to the Norwegian population, which was estimated to count 100,000–120,000 individuals in 1997 (Forchhammer et al. 1998; Langvatn 1998). An estimate of the ratio to the census size in red deer (Zachos et al. 2007), involved a present effective size of 25,000 individuals, divided between the demes identified through the STRUCTURE analyses. An equal ancient effective size was used in all scenarios to attain a similar and probable amount of genetic variation prior to demographic events. One common migration rate, estimated from the present genetic structure (F ST) identified by STRUCTURE and corresponding effective sizes, was used in all models. We simulated 14 microsatellite loci on independent chromosomes with a mutation rate of 10−4 and a geometrically distributed two-phase model (Di Rienzo et al. 1994) constrained to 20 alleles. For each scenario the effect of different effective deme sizes during the demographic events was modelled. For each model, 1,000 data sets were simulated and summary statistics calculated across these with Arlequin3.11 (Excoffier et al. 2005). The ranges of average gene diversity and F ST values among demes were then compared with our empiric red deer data divided according to the subpopulations identified through STRUCTURE.
Results
For each of the sampled localities, all loci were in Hardy–Weinberg equilibrium after sequential Bonferroni adjustment, except for the locus BM4208 in locality SE3 (P = 0.0004). We found a total of 74 alleles, an average gene diversity of H = 0.61 (SE = 0.02), an allelic richness of A = 4.1 (SE = 0.3), a F IS value of 0.018 (SE = 0.01) and an overall F ST value of 0.08 (SE = 0.02). We found significantly higher gene diversities than expected from the observed number of alleles in all of the localities except N1 (P = 0.09; Table 1), suggesting deviations from mutation-drift equilibrium after loss of alleles during recent bottlenecks. This was verified by the low M-ratio values in all localities, as a M-ratio smaller than 0.68 can be assumed to represent a recent reduction in population size (Garza and Williamson 2001). However, the M-ratio varied little between the localities, which had quite similar amounts of genetic variation (Table 1), with no differences in either allelic richness or gene diversity (One-way ANOVA; F = 0.24, F = 0.32, respectively; P = 0.99 for both parameters). Rather, the low number of observed alleles in the whole data set (74) compared to if all possible size repeat mutations had been present within the observed microsatellite repeat size range in all loci (142), suggests a general loss of alleles from the whole data set.
Among the 120 pairwise F ST-values between localities, 102 were significant after sequential Bonferroni correction, ranging from 0.004 to 0.188 (Table 2). Many of these indicated moderate (0.05–0.15) to strong (0.15–0.25) genetic structure (Wright 1978; Hartl and Clark 1997). Differentiation was particularly strong between the southernmost and northernmost localities along the coastline, and isolation-by-distance was highly significant (P < 0.0001). The NJ tree (Fig. 2) showed a main dichotomy between localities north and south of Sognefjorden, the largest fjord in Norway. The locality at Sognefjorden (W) showed an intermediate position in the NJ tree and was moderately, albeit significantly, differentiated from all other populations (Table 2). The south and south-eastern localities (S, SE 1–4) clustered with the south-western locality (SW) with high bootstrap values, indicating that these newly established localities were founded by dispersers from the southern part of the coastline. Similarly, the recently established eastern locality (E) clustered with the localities north on the coastline (N 1–4), indicating that its founders originated from the northern area.

Unrooted neighbour joining tree based on pairwise D A—distances among the 15 sampled localities. Bootstrap values above 50 are indicated (1,000 replicates)
The STRUCTURE algorithm showed that a partitioning of the genetic variation into five clusters was most probable when both independent and correlated allele frequencies were applied (P(K = 5|D) < 0.999). Moreover, a much higher variance among runs with K > 5 for the correlated allele frequencies model and for K > 6 for the independent allele frequencies model, indicate that five or six clusters represent the main genetic structure (Table 3). This was supported by a high delta value for K = 5 in the model with correlated allele frequencies. In both models an even higher delta value demonstrated a major break in the data set with K = 2, reflecting a dichotomy of genetic divergence between localities north and south of the Sognefjorden (W; Fig. 3), as could be expected from spatial expansion from the most differentiated localities. With K = 5, the proportionate cluster membership was for most of the localities much higher in one of the clusters (Table 4) and divides the data geographically into three clusters of localities along the north-western coast (clusters 1, 2, 3), one cluster from the south-western to the south-eastern coast (cluster 4) and one cluster in south-eastern to central Norway (cluster 5). Localities E and C, both newly established localities, had a strong affinity towards the northern and north-western areas. Fig. 3 shows the probabilities of individual assignment to each of the five clusters and visualises their geographic distribution. A large proportion of the individuals as well as localities have a genetic signature typical for one specific cluster. However, some individuals and localities have a divided membership between two and three clusters, indicating a mixed origin from different sources. Such a pattern is particularly pronounced in the south-eastern localities.

Individual posterior probabilities of Bayesian assignment to each of two to five clusters (different colours) among 419 red deer in each of 15 localities (separated by vertical lines) analysed by STRUCTURE with K ∈ [2,5]
The simulated demographic scenarios strongly indicate that the observed genetic structure is of a recent origin. The magnitude of genetic differentiation was much higher for the simulated bottlenecks within an ancient structure (model B) than in the empiric data, while relatively equal in most simulations of fragmentation (model A) and those of founding and merging events (model C) with low effective sizes in both source and sink demes (Table 5, Supplementary tables 1, 2). In addition did not empiric gene diversity vary between subpopulations when divided according to the STRUCTURE analyses, and equal gene diversity among demes resulted from simulations of fragmentation and of founder and merging events (C), even though both involved a higher level of variation, except for relatively low effective sizes. By comparison, the recent bottlenecks within an ancient structure involved too wide ranges among demes in both average gene diversity and pairwise F ST values to be comparable to the empiric values.
Discussion
Our analysis clearly showed that the Norwegian red deer is not a panmictic population. The many significant F ST values indicated limited gene flow among most of the sampled localities, especially between the northern and southern localities, and demonstrated the presence of moderate to strong genetic structure. We therefore reject our null hypothesis. We found that isolation-by-distance was significant among the localities, a pattern compatible with limited gene flow and random genetic drift within the localities. The STRUCTURE algorithm showed that a partitioning of the genetic variability into five clusters was most probable (99.9%), even though also indicating a higher hierarchical dichotomous breakpoint between localities north and south of Sognefjorden. Thus, we interpreted the expanding Norwegian red deer population to consist of five sub-populations, four distributed from north to south along the coast and the fifth situated in the central and south-eastern part of the sampled area.
Generally speaking, genetic structure in a spatially expanding population may result from both long distance dispersal and limited migration among demes (Nichols and Hewitt 1994; Ibrahim et al. 1996; Austerlitz et al. 1997; Excoffier 2004). The equal genetic variation among demes in both empiric data and the simulated founder and merging events (model C) supports this, but the lower level of genetic differentiation with this simulation model except with low sizes of source demes also indicates fragmentation (model A). In our particular case, the demographic history of the Norwegian red deer population may have played a significant role. After the population size had been reduced from the mid eighteenth century the Norwegian red deer were in the early twentieth century distributed among five or six main locations along the coast from the north to the south–west (Collett 1909; Ingebrigtsen 1924; Langvatn 1988). Four of these are concurrent with the four subpopulations we identified along the coastline, indicating that the observed genetic structure was formed by genetic drift during the population decline after the mid eighteenth century. Unfortunately, we did not sample the last one or two locations from this period. One of these, situated at the Bergen Peninsula, could be concurrent with our fifth STRUCTURE cluster. Indeed, three of the south-eastern localities (SE2, SE3, SE4) that are located close to the Bergen Peninsula had a high membership in this cluster suggesting partial foundation from this area. The comparable genetic variation and structure between the simulated fragmentation and the empiric data support that relict Norwegian localities became genetically differentiated through the recent population reduction and fragmentation. Founder events have probably also involved genetic differentiation, but may not account for the observed genetic structure among relict localities along the west coast.
The significant deviations from the heterozygosity expected with the observed number of alleles in 14 out of 15 localities suggested a recent bottleneck (Cornuet and Luikart 1996). Loss of alleles from a bottleneck was also indicated by the low M-ratio’s in all the localities. The Norwegian red deer population was abundant prior to the eighteenth century (Friis 1874; Collett 1877, 1909) and one likely period for a recent bottleneck of moderate magnitude was during the decline between the mid eighteenth and early twentieth century. Our simulations show that strong bottleneck events are unlikely to have happened recently since we in our empiric data did not record any corresponding differences in genetic variation. Since there was no such difference between old and recently established populations but rather similar bottleneck signals, these are probably not due to separate founder events but probably rather originates from one separate signal. This is supported by the low overall number of observed alleles compared to possible repeats in the allele size range of microsatellite loci, suggesting older and more severe bottlenecks prior to the abundant period in the 1500th and 1600th centuries than during the population reduction after the mid eighteenth century. Such a scenario is further supported by the lower gene diversity in the Norwegian red deer compared to the demographic simulations of both a recent fragmentation and of founding and subsequent merging events, suggesting a lower initial level before the onset of such demographic events. We conclude that the observed empiric genetic differentiation is probably recent rather than ancient and that the most likely explanation involves drift during the recent population reduction and fragmentation in combination with subsequent founder events during spatial expansion.
The recent expansion of the Norwegian red deer population has been drastic, both demographically and spatially, especially the last 50 years (Langvatn 1988; Forchhammer et al. 1998). By comparison, the neighbouring Swedish red deer population has not recovered to the same degree after the eighteenth and ninetieth century decline and still does not count more than 1,200–1,500 individuals in Scania (personal communication Anders Jarnemo, Swedish University of Agricultural Sciences). Analyses of the Swedish population (Vänersborg) indicated very low gene flow into the Norwegian population and assignment of individuals showed no admixture across the border (Haanes et al. unpublished). With the major population expansion the last century, we expected more gene flow and less present genetic structure in the primary area around the five relict populations. However, Southern Norway is divided by a central mountain range, which may constitute a barrier for dispersal from the west to the east. Moreover, the coastline is deeply punctuated by broad fiords with steep edges, constituting possible barriers for north–south dispersal along the coast. Accordingly, the significant F ST values between localities separated by fiords and inlets, like the island locality N2 and adjacent coastal localities (N3 and N4), indicated that water constitutes a barrier for red deer dispersal. Thus, even though long distance dispersal is common and red deer are frequently observed swimming (Collett 1909; Ingebrigtsen 1924; Langvatn 1988), our results showed that migration and dispersal along the coast have been limited. Similarly, the pattern of dispersal into the areas of new establishment seems to indicate that the massive mountain ranges of Norway have constituted barriers for red deer dispersal. The low genetic divergence between the south-western (SW) and south-eastern (SE) localities as well as between the north-western (NW) and eastern (E) localities demonstrate two main routes of range expansion, one from the area on the north-western coast towards south–east and one from the area on the south-western coast around the coastline and into south-eastern Norway. In addition, the close relationship between the central (C) and north-western (NW1, NW2) localities indicated foundation from the north-western coast by a third dispersal route across the northern part of the central Norwegian mountain range, where mountains are less alpine than further to the south. Dispersal from the area adjacent to Sognefjorden (W) seems to have been limited, presumably because the more massive and alpine mountains here functioned as a major barrier. The low degree of admixture between localities in the north, the south and the Sognefjord locality (W) further supported our interpretation of limited gene flow along the coast and across the highest mountain ranges of Norway. Equivalently, the high degree of admixture in the south–east and the central localities probably has been the result of higher migration and dispersal in this area which has fewer barriers of massive mountains and no large fiords with steep sides. Management could therefore take the identified barriers to dispersal into consideration and attempt to avoid genetic drift in the more isolated subpopulations on the west coast.
Conservation theory has traditionally been concerned with risk assessment of extinction in small or fragmented populations and which minimum population size that may avoid inbreeding and loss of genetic variation, and maintain adaptability (Lande 1988; Soulé and Mills 1992). The effects of subdivision on effective population size have been assessed in theoretic studies (Wang and Caballero 1999; Nunney 2000) and discussed in numerous empiric studies. Our data confirm that continued genetic loss during fragmentation may be counteracted by high levels of migration but even more importantly our simulations show that such differentiation may arise quite rapidly and not be as old as often interpreted. Our results thus support optimal management of fragmented populations through migration corridors to maintain gene diversity but also imply that structure often may be more recent than anticipated and not necessarily important to conserve.
The population density of Norwegian red deer is positively correlated to the North Atlantic Oscillations (Forchhammer et al. 1998, 2001; Mysterud et al. 2001). Thus, under a scenario of continued climatic change (IPCC 2001, 2007), we anticipate that the red deer population in Norway will continue to expand both demographically and spatially. The expanding parts from the north-western and the southern coast most likely will meet in the near future. Future studies of Norwegian red deer could include modelling of divergence times and may contribute even further to our understanding of the genetic effects of a spatial population expansion, where the whole process from complete isolation in small relict populations, through the expansion phase to a large population can be observed and analysed.
References
Ahlèn I (1965) Studies on the red deer, Cervus elaphus L. Scandinavia. III. Ecol investig Viltrevy (Stockh) 3:177–376
Austerlitz F, JungMuller B, Godelle B, Gouyon PH (1997) Evolution of coalescence times, genetic diversity and structure during colonization. Theor Popul Biol 51:148–164. doi:10.1006/tpbi.1997.1302
Baccus R, Ryman N, Smith MH, Reuterwall C, Cameron D (1983) Genetic variability and differentiation of large grazing mammals. J Mammal 64:109–120. doi:10.2307/1380756
Beaumont MA (1999) Detecting population expansion and decline using microsatellites. Genetics 153:2013
Begon M, Harper JL, Townsend CR (1996) Ecology, individuals, populations and communities, 3rd edn. Blackwell, Oxford
Bhebhe E, Kogi J, Holder DA et al (1994) Caprine microsatellite dinucleotide repeat polymorphism at the SR-CRSP-6, SR-CRSP-7, SR-CRSP-8, SR-CRSP-9 and SR-CRSP-10. Anim Genet 25:203
Bishop MD, Kappes SM, Keele JW et al (1994) A genetic linkage map for cattle. Genetics 136:619–639
Buchanan FC, Crawford AM (1993) Ovine microsatellites at the OarFCB11, OarFCB128, OarFCB193, OarFCB226 and OarFCB304 loci. Anim Genet 24:145
Chakraborty R, Nei M (1977) Bottleneck effects on average heterozygosity and genetic distance with stepwise mutation model. Evol Int J Org Evol 31:347–356. doi:10.2307/2407757
Chakraborty R, Kimmel M (1999) Statistics of microsatellite loci: estimation of mutation rate and pattern of population expansion. In: Goldstein DB, Schlotterer C (eds) Microsatellites; evolution and applications. Oxford University Press, Oxford, pp 139–150
Collett R (1877) Bemerkninger til Norges Pattedyrfauna (in Norwegian). Nyt Magazin for Naturvidenskaberne 22:93–133
Collett R (1909) Hjorten i Norge (Cervus elaphus atlanticus), nogle biologiske meddelelser (in Norwegian). Bergens museums Aarbok 6:9–31
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Di Rienzo A, Peterson AC, Garza JC et al (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170. doi:10.1073/pnas.91.8.3166
Ede AJ, Pierson CA, Crawford AM (1995) Ovine microsatellites at the OarCP9, OarCP16, OarCP20, OarCP21, OarCP23 and OarCP26 loci. Anim Genet 25:129–130
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839. doi:10.1007/BF00221895
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13:853–864. doi:10.1046/j.1365-294X.2003.02004.x
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: An integrated software package for populations genetics data analysis. Evol Bioinform Online 1:47–50
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Forchhammer MC, Clutton-Brock TH, Lindstrøm J, Albon SD (2001) Climate and population density induce long-term cohort variation in a northern ungulate. J Anim Ecol 70:721–729. doi:10.1046/j.0021-8790.2001.00532.x
Forchhammer MC, Stenseth NC, Post E, Langvatn R (1998) Population dynamics of Norwegian red deer: density-dependence and climatic variation. Proc R Soc Lond B Biol Sci 265:341–350. doi:10.1098/rspb.1998.0301
Friis JA (1874) Tilfjelds i ferierne (in Norwegian). Cammermeyer, Christiania
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318. doi:10.1046/j.1365-294x.2001.01190.x
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices. Release 2.9.3.2. Available from http://www.unil.ch/izea/softwares/fstat.html
Gyllensten U, Ryman N, Reuterwall C, Dratch P (1983) Genetic differentiation in four European subspecies of red deer (Cervus elaphus L.). Heredity 51:561–580. doi:10.1038/hdy.1983.71
Haanes H, Rosef O, Veiberg V, Røed KH (2005) Microsatellites with variation and heredity applicable to parentage and population studies of Norwegian red deer (Cervus elaphus atlanticus). Anim Genet 36:454–455. doi:10.1111/j.1365-2052.2005.01343.x
Hartl DL, Clark AG (1997) Principles of population genetics, 3rd edn. Sinauer, Sunderland, US
Hedrick PW (2000) Genetics of populations. Jones and Bartlett, Boston
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. doi:10.1038/35016000
Hewitt GM (2001) Speciation, hybrid zones and phylogeography—or seeing genes in space and time. Mol Ecol 10:537–549. doi:10.1046/j.1365-294x.2001.01202.x
Hulme DJ, Silk JP, Redwin JM, Barendse W, Beh KJ (1994) Ten polymorphic ovine microsatellites. Anim Genet 25:434–435
Ibrahim KM, Nichols RA, Hewitt GM (1996) Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77:282–291. doi:10.1038/hdy.1996.142
Ingebrigtsen O (1924) Hjortens utbredelse i Norge (in Norwegian). Bergens Museums Aarbok 1922–1923 Naturvitensk. Raekke 6:1–58
IPCC (2001) Third assessment report of the Intergovernmental Panel on Climate Change. Cambridge Univ Press, Cambridge
IPCC (2007) Fourth assessment report of the Intergovernmental Panel on Climate Change. http://www.ipcc.ch
Lande R (1988) Genetics and demography in biological conservation. Science 241:1455–1460. doi:10.1126/science.3420403
Langvatn R (1988) Hjortens utbredelse i Norge-en oversigt. Villreinen 1:1–8 in Norwegian
Langvatn R (1998) Hjortens erobring av Norge. In: Brox KH (ed) Brennpunkt natur (in Norwegian). Tapir Cop, Trondheim, pp 49–71
Laval G, Excoffier L (2004) SIMCOAL 2.0: a program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics 20:2485–2487. doi:10.1093/bioinformatics/bth264
Lønnberg E (1906) On the geographic races of red deer in Scandinavia. Arkiv för Zoologi 3:1–19
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Moore SS, Byrne K, Berger KT et al (1994) Characterization of 65 bovine microsatellites. Mamm Genome 5:84–90. doi:10.1007/BF00292333
Mysterud A, Stenseth NC, Yoccoz NG, Langvatn R, Steinheim G (2001) Nonlinear effects of large-scale climatic variability on wild and domestic herbivores. Nature 410:1096–1099. doi:10.1038/35074099
Mysterud A, Langvatn R, Yoccoz NG, Stenseth NC (2002) Large-scale habitat variability, delayed density effects and red deer populations in Norway. J Anim Ecol 71:569–580. doi:10.1046/j.1365-2656.2002.00622.x
Nei M et al (1975) The bottleneck effect and genetic variability in populations. Evol Int J Org Evol 29:1–10. doi:10.2307/2407137
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, NY
Nei M (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol 19:153–170. doi:10.1007/BF02300753
Nichols RA, Hewitt GM (1994) The genetic consequences of long-distance dispersal during colonization. Heredity 72:312–317. doi:10.1038/hdy.1994.41
Nunney L (2000) The limits to knowledge in conservation genetics; The value of effective population size. In: Clegg MT, Hecht MK, MacIntyre RJ (eds) The limits to knowledge in conservation genetics. Kluwer, NY, pp 179–194
Page RDM (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20:76–86. doi:10.1093/molbev/msg009
Raymond M, Rousset F (1995) GENEPOP (Version-1.2)—population-genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evol Int J Org Evol 43:223–225. doi:10.2307/2409177
Røed KH (1998) Microsatellite variation in Scandinavian Cervidae using primers derived from Bovidae. Hereditas 129:19–25. doi:10.1111/j.1601-5223.1998.00019.x
Røed KH, Midthjell L (1998) Microsatellites in reindeer, Rangifer tarandus, and their use in other cervids. Mol Ecol 7:1773–1778
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial-DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Soulé ME, Mills S (1992) Conservation genetics and conservation biology: a troubled marriage. In: Sandlund OT, Hindar K, Brown AHD (eds) Conservation of biodiversity for sustainable development. Scandinavian University Press, Oslo, pp 55–69
Wang JL, Caballero A (1999) Developments in predicting the effective size of subdivided populations. Heredity 82:212–226. doi:10.1038/sj.hdy.6884670
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer, Sunderland
Wilson GA, Strobeck C, Wu L, Coffin J (1997) Characterization of microsatellite loci in caribou Rangifer tarandus, and their use in other artiodactyls. Mol Ecol 6:697–699. doi:10.1046/j.1365-294X.1997.00237.x
Wright S (1978) Evolution and the genetics of populations. University of Chicago Press, Chicago
Zachos F, Althoff C, Steynitz Y, Eckert I, Hartl G (2007) Genetic analysis of an isolated red deer (Cervus elaphus) population showing signs of inbreeding depression. Eur J Wildl Res 53:61–67. doi:10.1007/s10344-006-0065-z
Acknowledgments
For help providing samples we thank the section for wildlife diseases at the Norwegian national veterinary intitute, Dr. Jon M. Arnemo, Harald Holm, M. Pearson, Halvor Ovastrøm, Oddegeir Hårstad, the hunters that sent us samples, and the game managers in the counties and municipalities of Norway who organised much of the sampling. For help handling samples and in the laboratory we are in debt to Turid Vikøren, Astrid Stovner and Liv Midthjell. For helpful information about the SIMCOAL algorithm and its function we thank Dr. Christian N. K. Anderson at the department of biological sciences, Stanford University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Haanes, H., Røed, K.H., Flagstad, Ø. et al. Genetic structure in an expanding cervid population after population reduction. Conserv Genet 11, 11–20 (2010). https://doi.org/10.1007/s10592-008-9781-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-008-9781-0