
Abstract
Genetic variations in the giant panda populations in Wanglang and Baoxing Nature Reserves were evaluated in this study. Panda feces were collected from these two reserves and DNA samples extracted from the feces were genotyped at 13 microsatellite loci. A total of 130 alleles were identified from the 13 microsatellite loci in 63 giant pandas, including 35 private alleles in Wanglang, 53 private alleles in Baoxing, and 42 alleles shared between the two populations. The mean observed heterozygosity, average number of alleles, average number of allelic richness, and average polymorphism information content were 0.488, 6.2, 3.302, and 0.612, respectively for the Wanglang population and 0.553, 7.6, 4.050, and 0.747 for the Baoxing population. A moderate degree of genetic differentiation (F st = 0.26) and no gene flow were found between these two populations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The giant panda (Ailuropoda melanoleuca) is one of the most critically endangered species in the world with a total of 1,600 individuals reported in the latest survey. Historically, this species was widely distributed in southwestern China, but is now restricted to six isolated mountain ranges along the eastern edge of the Tibetan Plateau. The range has been fragmented into more than 30 isolated habitats and the population divided into an estimated 24 groups, most with less than 50 individuals (O’Brien et al. 1994; Loucks et al. 2001). These small, isolated populations face a great risk of inbreeding depression.
In previous studies, various kinds of genetic markers were used to determine the genetic diversity of giant pandas. Protein electrophoresis showed that the percentage of polymorphic loci and mean individual heterozygosity were extremely low (Su et al. 1994). Sequence analysis of the mitochondrial control region indicated a low genetic diversity with a comparatively high haplotype (Zhang et al. 1997). At the same time, Wan et al. (2006) found relatively low numbers of alleles of the MHC class II DRB locus in giant pandas. However, Lu et al. (2001) concluded that panda populations still retained considerable genetic diversity when compared against other carnivorous species, which was insured by Wan et al. (2003). Fang et al. (2002, 2003) found the genetic variation in giant panda populations has a relationship with the number of individuals and that larger populations possessed relatively higher genetic variation than smaller populations. Wan et al. (2003, 2005) indicated that the Qinling population had diverged into a new subspecies (A. m. qinlingensis) when compared to the original subspecies (A. m. melanoleuca) in Sichuan. These differences showed that further systematic studies were required to clarify the genetic situation of the giant panda in the wild.
Microsatellite markers are widely applied in genetic variation studies on endangered species because of the high levels of variations, tiny amount of DNA required and noninvasive sampling techniques (Jarne and Lagoda 1996; Beaumont and Bruford 1999). Wanglang and Baoxing Nature Reserves are among the most important national reserves for giant pandas in Sichuan Province. Wanglang Nature Reserve is located in the northern Minshan Mountains and is where the largest giant panda population occurs. It was established in 1965 and is an important habitat corridor for pandas. There are almost 140 individuals living in Baoxing Nature Reserve, representing more than 10% of the total population (http://unn.people.com.cn/GB/14796/21796/2735558.html). However, few studies have reported the levels of genetic diversity for these two wild populations. The aim of this study was to evaluate the genetic variations within and between these two wild panda populations.
Materials and methods
Sample collection and DNA preparation
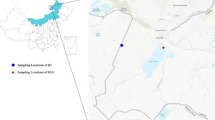
Wild giant panda fecal samples were collected from Wanglang Nature Reserve and Baoxing Nature Reserve in 2005. In order to reduce the number of samples taken from the same individual, another sample could not be collected for 3 km after the previous sampling, as determined from home range studies (Hu 1990). All sampling positions were recorded by GPS and mapped using Arcview 3.2a (Fig. 1). Up to 5 g of feces were peeled from the outer layer and stored in 95% ethanol. Genomic DNA was extracted using the QIAamp® Stool Mini Kit (Qiagen) under the manufacturer’s instructions (Standard Qiagen Technique).

Sketch map of sample points in Baoxing and Wanglang
DNA quality verification
A total of 121 DNA samples were verified by PCR amplifications using three microsatellite loci: Panda-40, gp901 and Ame-μ25 (Table 1). DNA samples with at least one successful amplification in those three loci were considered eligible for further microsatellite amplifications. Otherwise, the samples were not used.
Microsatellite analysis
A total of 13 microsatellite loci (Table 1) originally designed in giant pandas were amplified to analyze the genetic structure of the two wild populations under previously published conditions (Zhang et al. 1995, 2003; Shen et al. 2007;). Primers were fluorescently labeled with 5′-FAM or TET. Allele sizes were determined by separating the amplification products on a DNA sequencing and genotyping gel (Cambrex Bio Science Rockland) and run on an ABI 373 DNA Analyser (Applied Biosystems). Fragment length was determined by Genescan software (Applied Biosystems) using the GeneScan-350 (TAMRA) size standard.
Because very dilute fecal DNA was used in this study, the “Multi-tube Procedure” was employed to avoid genotyping errors due to potential allelic dropout (Taberlet et al. 1996). The 13 microsatellite loci were amplified for seven replications on each of the 63 DNA samples and only those loci with 3 or more successful PCR amplifications in the seven replicates were used for genotyping analysis. Based on the “Multi-tube Procedure” guidelines, when two alleles for each locus were amplified unambiguously three or more times in the seven replicates on a given DNA sample, the sample was considered heterozygous, even if the two alleles appeared in different amplifications. Only one allele for each locus would be amplified in the seven PCR replications if it were homozygous. More details for other situations were described by Taberlet et al. (1996).
Data analysis
Genotypes from different samples were considered to represent the same individual if identical alleles were found on 12 or more loci (Bellemain et al. 2005). Genetic polymorphism was estimated for each population as the number of alleles per locus (A), observed heterozygosity (H o), expected heterozygosity (H e), and polymorphic information content (PIC) using the program CERVUS 2.0 (Marshall et al. 1998).
GENEPOP 3.3 (Raymond and Rousset 1995) was used to test the genotypic distribution for conformance with Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD). All probability tests were based on the Markov chain method using 1,000 de-memorization steps, 100 batches and 1,000 iterations per batch. Allelic richness (A R) was calculated with the FSTAT 2.9.3 program package (Goudet 2001). Allele frequency and private alleles were analyzed through the software CONVERT 1.31 (Glaubitz 2004). An assessment of population pairwise comparisons was derived from ARLEQUIN 3.1 and the statistical significance of F st values was tested with 10,000 permutations as implemented in ARLEQUIN 3.1 (Excoffier et al. 2005).
To detect the genetic signature of a population decline, the soft program BOTTLENECK and a two-phased model of mutation (TPM) were applied (Piry et al. 1999). The Wilcoxon sign-rank test was used to determine significance. The marginal probability of each individual in each population was calculated, based on the allele frequencies of the samples, and the individual was assigned to the population in which it had the highest marginal probability. A Bayesian clustering method was used to infer population structure based upon the software STRUCTURE (Pritchard et al. 2000).
Results
Genetic diversity and bottleneck analysis
A total of 121 stool samples from Wanglang and 51 from Baoxing were collected (Fig. 1). However, only 33 DNA samples from Wanglang and 30 from Baoxing were considered suitable for further microsatellite analysis based on quality verifications. Microsatellite genotyping analysis revealed that these 63 samples included 63 unique genotypes and were from 63 different individuals. The percent of lacking genotypes was 21.8% in the 63 samples and 13 loci (Table 1). The frequency of false homozygotes (FH) ranged from 0% (such as the Panda-40 locus) to 18% (Panda-05) depending on the locus, with an average of 10%. False alleles (FA) and multiple alleles (MA) were not found in our study.
A total of 130 alleles were identified for the 13 microsatellite loci in the 63 samples (Table 1), including 35 private alleles in Wanglang, 53 private alleles in Baoxing and 42 alleles shared between the two populations. The number of alleles per locus ranged from six at locus gp03 to a maximum of 16 at locus Panda-05 (Table 1). When compared to genetic diversity indices for the Wanglang population, there was a higher genetic diversity in the Baoxing population based on the higher average number of allelic richness (A R = 4.05) (Table 1), but the difference was not significant (P = 0.388).
Populations experiencing a recent bottleneck would display a reduction in allele number and, more slowly, heterozygosity. Excess heterozygosity in a population at mutation-drift equilibrium could therefore indicate a recent population decline (Cornuet and Luikart 1996). No evidence for recent bottlenecks in these two populations was found based on the Wilcoxon sign-rank test (P < 0.05) using the infinite allele model (IAM), two-phase model (TPM) and stepwise mutation model (SMM).
Linkage disequilibrium (LD), Hardy–Weinberg equilibrium heterozygosity
Following Bonferroni correction, highly significant LD tests were not shown for any pair of loci within each of the populations. Hardy–Weinberg equilibrium (HWE) tests revealed that seven loci in the Wanglang population (P < 0.004) and six loci in the Baoxing population (P < 0.03) deviated from HWE (Table 1). To test whether nonrandom mating was responsible for departure from HWE for these loci, we calculated F is and found that F is ranged from 0.816 to 0.103 in the Wanglang population and 0.700–0.254 in the Baoxing population. The significant heterozygote deficiency (P < 0.02) in these loci was probably the reason for departure from HWE.
A large variation in heterozygosity was observed in this study. The mean expected heterozygosity (He) was 0.680 (ranging from 0.169 to 0.967) in Wanglang, as compared to 0.819 (ranging from 0.577 to 0.935) in Baoxing and the mean observed heterozygosity was 0.488 in Wanglang and 0.553 in Baoxing (Table 1). All observed heterozygosities (H o) were lower than the corresponding expected heterozygosities, except for loci Ame-μ15 and gp01. For some loci, the observed heterozygosity varied greatly between the two populations, such as Ame-μ21 and Ame-μ25 (Table 1). The mean F is values were 0.29 for Wanglang and 0.32 for Baoxing.
Genetic differentiation between the two populations
In the genetic differentiation analysis, only 29 individuals (16 from Wanglang and 13 from Baoxing) were used because some fecal samples amplified successfully on less than nine of the 13 microsatellite loci. The results indicate a moderate degree of genetic differentiation between the Baoxing and Wanglang populations with an F st-value of 0.26 (P < 0.0001). In assignment tests, when these 29 individuals were assigned to two clusters (when K = 2, the probability of the data in the Ln probability was the highest.) using the Bayesian clustering method in STRUCTURE (Pritchard et al. 2000), all individuals clustered clearly to the population where they came from and we could not identify any immigrant individual from another population (Fig. 2). These populations could be considered to be genetically distinct and their uniqueness was supported by private alleles as well.

Bayesian Clustering Analyses through the software STRUCTURE (admixture model, burn-in and replication values set at 50,000 and 106, respectively). Black circle: Wanglang; Gray circle: Baoxing
Discussion
Panda feces is one of the most important sources of information for genetic and ecological research and many studies show that microsatellite amplifications can be successfully applied using a tiny amount of DNA obtained from feces (Zhan et al. 2006). However, sometimes it was very difficult to get high quality DNA from feces collected from the wild. In our study, we sampled 172 panda feces and obtained only 63 good quality DNA samples for microsatellite amplifications. This was mostly because of degradation caused by remaining in the environment for a long period of time prior to sampling or improper storage. Fecal materials for our study were collected by staff in the nature reserves and some samples were estimated to have remained in the wild for over a month. Secondly, some sampled feces in ethanol were maintained at room temperature for over a month before being delivered to our laboratory for DNA preparations because there was no refrigerator at the reserves. Therefore, in order to obtain a higher quality DNA, fresh feces (less than 2 weeks) must be used and the DNA extracted as soon as possible after sampling.
DNA quality verification before formal amplifications is very important for noninvasive samples. In our study, three microsatellite loci were used to verify the 172 samples collected from Wanglang and Baoxing. Following verification, only 63 of the 172 DNA samples were considered suitable for genetic structure analysis using the 13 microsatellite loci. In this way, many unnecessary extra amplifications were avoided.
In theory, heterozygote deficiencies could result from inbreeding, selection against heterozygotes, the Wahlund effect (population subdivision into separate breeding units), underdominant selection, presence of null alleles, and missing data (Sun and Salomon 2003). Our studies suggest that a deficit of heterozygotes in the two populations may be caused by inbreeding and missing data. The reason for the deficit also might be due to the presence of null alleles in some individuals as Shen et al. (2007) found that the flanking sequence of some microsatellite loci have high mutation rates in giant panda. The missing genotypes were randomly distributed in the dataset and therefore likely to be random occurrences due to failed PCR amplifications. Our microsatellite genotyping protocol strictly followed the stringent criteria of Taberlet et al. (1996), which could produce reliable genotypes at a 99% confidence level for characterizing homozygous individuals. The mean F is value was 0.29 for Wanglang and 0.32 for Baoxing and therefore inbreeding was also probably a cause of heterozygote deficiency.
A very low genetic differentiation (F st = 0.07; P > 0.05) was reported for panda populations in Minshan Mountains and Qionglai Mountains (Lu et al. 2001). Wan et al. (2003) suggested that the giant pandas in these two areas should be managed together to increase their gene flow. However, a moderate population differentiation (F st = 0.26; P < 0.0001) was found between the Wanglang and Baoxing populations in our study. This conclusion was consistent with the results obtained by Zhan et al. (2006). At the same time, this differentiation was also shown in the assignment tests. All individuals from the same population were clustered together and this result revealed that no gene flow existed between these two populations (Fig. 2). Using the high information content of multiple hypervariable microsatellite loci, assignment tests are increasingly recognized as a novel method in wildlife management, conservation genetics and forensics (Luikart and England 1999; Primmer et al. 2000; Eldridge et al. 2001). The assignment process has recently been refined with the development of a simulation procedure which determines the probability of an individual originating from a series of potential source populations (Cornuet et al. 1999).
Changes in allelic diversity probably occur faster in isolated populations and therefore cause stronger differences between those populations. Maruyama and Fuerst (1985) predicted that allelic variation probably declines faster than heterozygosity. This was found in fragmented populations of tamarins, Leontopithecus rosalia, in Brazil (Grativol et al. 2001). Wild giant panda populations are facing serious habitat fragmentation and loss of allelic diversity would be as serious a concern. Fortunately, low inbreeding (the mean F is values were 0.29 for Wanglang and 0.32 for Baoxing) and no recent population bottlenecks were found in the Wanglang and Baoxing panda populations and the average heterozygosity was higher than in cheetahs (0.443), pumas (0.357) and lions (0.373) (Lu et al. 2001). It seems, therefore, that the giant panda populations in Wanglang and Baoxing have the potential to be restored through our efforts.
References
Beaumont MA, Bruford MW (1999) Microsatellites in conservation genetics. In: Goldstein DB, Schlötterer C (eds) Microsatellites: evolution and applications. Oxford University Press, pp 165–182
Bellemain E, Swenson JE, Tallmon D, Brunberg S, Taberlet P (2005) Estimating population size of elusive animals with DNA from hunter-collected feces: four methods for brown bears. Conserv Biol 19:150–161
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M (1999) New methods employing multi-locus genotypes to select or exclude populations as origins of individuals. Genetics 153:1989–2000
Eldridge MDB, Kinnear JE, Onus ML (2001) Source population of dispersing rock-wallabies (Petrogale lateralis) identified by assignment tests on multilocus genotypic data. Mol Ecol 10:2867–2876
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fang SG, Wan QH, Fujihara N (2002) Genetic diversity of the giant panda (Ailuropoda melanoleuca) between big and small populations. J Appl Anim Res 21:65–74
Fang SG, Wan QH, Fujihara T (2003) Loss of genetic variation in giant panda due to limited population and habitat fragmentation. J Appl Anim Res 24:137–144
Glaubitz JC (2004) Convert: a user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes 4:309–310
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html. Updated from Goudet J (1995) FSTAT v-1.2. A computer program to calculate F-statistics. J Hered 86:485–486
Grativol AD, Ballou JD, Fleischer RC (2001) Microsatellite variation within and among recently fragmented populations of the golden tamarin (Leontopithecus rosalia). Conserv Genet 2:1–9
Hu J (1990) The biological studies of the giant panda. In: Hu J, Wei F, Yuan C, Wu Y (eds) Research and progress in biology of giant panda. Sichuan Public House of Science and Technology, Chengdu, China, pp 24–28
Jarne P, Lagoda PJ (1996) Microsatellites, from molecules to populations and back. Trends Ecol Evol 11(10):424–429
Loucks CJ, Lu Z, Dinerstein E, Wang H, Olson DM, Zhu CQ, Wang D (2001) Giant pandas in a changing landscape. Science 294(16):1465
Lu Z, Johnson WE, Menotti-Raymond M, Yuhki N, Martenson JS, Mainka S, Huang S-Q, Zhang Z-H, Li G-H, Pan W-S, Mao X-R, O’Brien SJ (2001) Patterns of genetic diversity in remaining giant panda populations. Conserv Biol 15(6):1596–1607
Luikart G, England PR (1999) Statistical analysis of microsatellite DNA data. Trends Ecol Evol 14:253–256
Marshall TC, Slate J, Kruuk L, Pemberton JM (1998) Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7:639–655
Maruyama T, Fuerst PA (1985) Population bottlenecks and non-equilibrium models in population genetics. II. Number of alleles in small population that was formed by a recent bottleneck. Genetics 111:675–689
O’Brien SJ, Pan WS, Lu Z (1994) Pandas, people and policy. Nature 369:179–180
Piry S, Luikart G, Cornuet JM (1999) Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503
Primmer CR, Koskinen MT, Piironen J (2000) The one that did not get away: individual assignment using microsatellite data detects a case of fishing competition fraud. Proc R Soc Lond B Biol Sci 267:1699–1704
Pritchard JK, Stephens M, Donelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Shen F, Watts PC, He W, Zhang Z, Zhang A, Sanderson S, Kemp SJ, Yue B (2007) Di-, tri- and teranucleotide microsatellite loci for the giant panda, Ailuropoda melanoleuca. Mol Ecol Notes. doi:10.1111/j.1471-8286.2007.0185.x
Su B, Shi L, He G (1994) The genetic diversity research of giant panda through protein electrophoresis. Chin Sci Bull 39(15):1305–1307 (in Chinese)
Sun G, Salomon B (2003) Microsatellite variability and heterozygote deficiency in the arctic–alpine Alaskan wheatgrass (Elymus alaskanus) complex. Genome 46:729–737
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194
Wan QH, Fang SG, Wu H (2003) Genetic differentiation and subspecies development of the giant panda as revealed by DNA fingerprinting. Electrophoresis 24:1353–1359
Wan QH, Wu H, Fang SG (2005) A new subspecies of giant panda (Ailuropoda melanoleuca) from Shaanxi, China. J Mammal 86:397–402
Wan QH, Zhu L, Wu H, Fang SG (2006) Major histocompatibility complex class II variation in the giant panda (Ailuropoda melanoleuca). Mol Ecol 15:2441–2450
Zhan X, Li M, Zhang Z, Goossens B, Chen Y, Wang H, Bruford MW, Wei F (2006) Molecular censusing doubles giant panda population estimate in a key nature reserve. Curr Biol 16:R451–R452
Zhang Y, Ryder OA, Fan Z, Zhang H, He T, Zhang A, Fei L, Zhong S, Chen H, Zhang C, Yan M, Zhu F, Peng Z, Pu T, Chen Y, Yao M, Guo W (1997) DNA sequences and genetic diversity of giant panda. Sci China C 27(2):139–144 (in Chinese)
Zhang Y-P, Wang W, Su B, Ryder OA, Fan Z-Y, Zhang H-M, He T-M (1995) The filter and apply of giant panda microsatillite DNA. Zool Res 16(4)301–306 (in Chinese)
Zhang Z-H, Shen F-J, Sun S, Daivid VA, Zhang A-J, O’Brien SJ (2003) Use microsatillite to identify the father of giant panda. Genetics 25(5):504–510 (in Chinese)
Acknowledgements
The authors would like to thank Churmy Fan and Yingting Luo for many useful suggestions on the manuscript. We would also like to thank the reserve staff for collecting panda stool samples. This research was Financially Supported by the National Basic Research Program of China (973 project: 2007CB411605) and the Chengdu Giant Panda Breeding Research Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
W. He and L. Lin contributed equally to this work.
Rights and permissions
About this article
Cite this article
He, W., Lin, L., Shen, F. et al. Genetic diversities of the giant panda (Ailuropoda melanoleuca) in Wanglang and Baoxing Nature Reserves. Conserv Genet 9, 1541–1546 (2008). https://doi.org/10.1007/s10592-007-9493-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-007-9493-x