
Abstract
Endangered species worldwide exist in remnant populations, often within fragmented landscapes. Although assessment of genetic diversity in fragmented habitats is very important for conservation purposes, it is usually impossible to evaluate the amount of diversity that has actually been lost. Here, we compared population structure and levels of genetic diversity within populations of spotted suslik Spermophilus suslicus, inhabiting two different parts of the species range characterized by different levels of habitat connectivity. We used microsatellites to analyze 10 critically endangered populations located at the western part of the range, where suslik habitat have been severely devastated due to agriculture industrialization. Their genetic composition was compared with four populations from the eastern part of the range where the species still occupies habitat with reasonable levels of connectivity. In the western region, we detected extreme population structure (F ST = 0.20) and levels of genetic diversity (Allelic richness ranged from 1.45 to 3.07) characteristic for highly endangered populations. Alternatively, in the eastern region we found significantly higher allelic richness (from 5.09 to 5.81) and insignificant population structure (F ST = 0.03). As we identified a strong correlation between genetic and geographic distance and a lack of private alleles in the western region, we conclude that extreme population structure and lower genetic diversity is due to recent habitat loss. Results from this study provide guidelines for conservation and management of this highly endangered species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Population fragmentation and isolation may have detrimental effects both on the fitness and viability of extant populations (Frankham et al. 2002). Local bottlenecks can reduce genetic variation, thus compromising the ability of a population to respond to environmental change (Amos and Balmford 2001). Numerous papers describe the effects of habitat loss on changes of genetic composition of populations, from Western Europe, North America, Australia or places identified as biodiversity hotspots. However, results of habitat degradation are already detectable also in the less industrialized regions of Eastern Europe (Honnay et al. 2007; Rosenbaum et al. 2007; Barnett et al. 2006; Ramirez et al. 2006). This study, concerning the effect of habitat fragmentation on the genetic diversity of critically endangered spotted suslik Spermophilus suslicus, is one of the first examples of using molecular markers in planning the conservation management programme in Eastern Europe. Species with low dispersal ability, like rodents, are especially sensitive to the effects of habitat fragmentation. Hirota et al. (2004) proved higher levels of population structure in Japanese field mouse populations from fragmented suburban areas in comparison to more continuous habitat. Lowered genetic diversity was also found in two subspecies of Idaho ground squirrel Spermophilus brunneus (Garner et al. 2005).
Spotted suslik is a medium sized rodent species that inhabits dry meadows, steppes and pastures of the Ukraine, Moldova, the European part of Russia and Poland. Polish populations are the westernmost populations of this species. Over the last 50 years, rapid development of agriculture and industrialization has led to an exceptionally fast loss of semi-natural habitats, such as extensively used meadows or pastures, which in turn caused a dramatic reduction in both distribution and abundance of spotted suslik in the western part of its range. Most populations became extinct and the extant ones became severely depleted in numbers and separated by intensively used agricultural landscapes. Presently, spotted suslik is considered as one of the rarest mammal species in Poland. Out of the 143 colonies noted in 1954 (Surdacki 1963), there are only seven left (Gondek 2004). The species is strictly protected under Polish and Bern Convention Laws and is listed as endangered in the Polish Red Data Book, under IUCN categories (Głowaciński and Męczyński 2001). Spotted suslik is also listed in the EU Habitat Directive and, importantly, Polish populations of this species are the only colonies in the European Union.
The assessment of the loss of variability is often hampered by a lack of information, about the genetic variation of threatened populations prior to fragmentation and demographic bottlenecks. The comparison of the pattern of genetic diversity before and after environmental or demographic perturbances is usually not possible. While Polish populations of spotted suslik are restricted to small, remnant, isolated colonies, the eastern part of the species range still sustains many small colonies densely distributed across suitable habitat (Lobkov 1999), providing a rare opportunity to investigate the natural processes shaping the population structure and gene flow among populations and to compare them with populations strongly affected by habitat deterioration.
In this study, we used microsatellite markers to investigate genetic diversity, population genetic structure and gene flow among and within spotted suslik colonies in two parts of its range, that differ in terms of levels of habitat connectivity and suitability. We compared the measures of population diversity and population structure of threatened populations, from the western part of the species range located in Poland and western Ukraine, with the diversity of populations from the main range situated in south-eastern Ukraine. We predicted that due to barriers to dispersal and recent extinctions of neighbouring populations in the western region, distances between populations increased, and consequently gene flow between remaining populations is likely to be seriously reduced or even absent. Combined with low population sizes and known size bottlenecks, due to deteriorated habitat quality and historical processes typical for peripheral populations, spotted suslik may suffer from decreased levels of genetic diversity and elevated levels of population structure.
Materials and methods
Sample collection
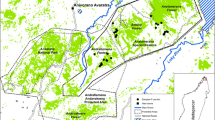
During the years 2003–2005, tip tail tissue samples were collected from 251 individuals of spotted suslik from 14 populations, covering two separate regions from the species range. We sampled six populations from the western region located in eastern Poland, in the area of the town of Zamość and four located in the neighbouring area in Ukraine, about 10–15 km from the Polish border. Another four populations located near the town of Odessa represented the eastern region. Sampling details are presented on Fig. 1 and in Table 1. Field procedures were carried out according to the Ethical Committee decision no.: 48/OP/2002 and the Polish Ministry of Environment permit no.: DOPog–4201-04-73/03/jr and DOPog–4201-04-22/04/jr). Individuals were captured in wire-mesh tubes placed at the entrance of burrows. The traps were spread evenly throughout the area to avoid non-random sampling of related individuals.

Geographic distribution of sampled spotted suslik populations; abbreviations as in Table 1
DNA extraction and microsatellite typing
Genomic DNA was extracted from ethanol-preserved tail tips. Samples were dried in 70°C for at least 5 h and the DNA was extracted by Nucleospin Tissue Kit (Macherey and Nagel, Germany) according to the manufacturer’s instructions. Individual samples were genotyped at nine microsatellite loci described in Gondek et al. (2006) and two additional loci developed for Marmota marmota (locus MS53, Hanslik and Kruckenhauser 2000) and Spermophilus brunneus (locus BP1, May et al. 1997). PCR reactions were carried out in a PTC-200 thermocycler (MJ Research). All PCR reactions were performed in a 10 μl volume containing 1× PCR buffer with (NH4)2SO4 (Fermentas), 2.0 mM MgCl2, 0.2 mM of each dNTP, 2 pmol of each primer, 0.5 U of Taq polymerase (Fermentas) and 5 ng of template DNA. One primer of each pair was fluorescent dyed. PCR conditions were as described in Gondek et al. (2006).
Amplification products were resolved on an ABi Prism 310 Genetic Analyser (Applied Biosystems, USA).
Data analysis
Tests for Hardy–Weinberg equilibrium HWE and linkage equilibrium between all pairs of loci for all the populations sampled and for all the data pooled together, were completed with the FSTAT (Goudet 2002) software. The deviations from HWE were expressed as an inbreeding coefficient F IS at each locus in each population and the significance was determined through permutation. The significance of linkage equilibrium was assessed with exact test and permutation scheme. Resultant P-values were corrected for the number of tests via sequential Bonferroni correction and significance level was adjusted for 0.05. Standard diversity indices have been calculated for each population. Observed and expected heterozygosities for each population were calculated using the FSTAT (Goudet 2002). Since allelic diversity is strongly affected by sample size, allelic richness (R) and private allelic richness (R priv) were obtained with the HP-rare (Kalinowski 2005) software. Corrected estimates of allelic richness and private allelic richness were based upon the smallest sample size (n = 5). To compare the levels of genetic diversity from the western, “fragmented” and eastern “continuous” populations, we analyzed the means of observed and expected heterozygosity, inbreeding coefficient F IS, allelic richness and relatedness coefficient (Queller and Goodnight 1989), for both regions using the FSTAT option for comparing groups of samples. For this purpose average indices for each group, over samples and loci, were calculated. The significance was assessed through permutation scheme as implemented in FSTAT. Whole samples were allocated at random to different groups and P-value of the test was taken as the proportion of randomized datasets giving a value larger than observed. To test if the difference in population structure between regions originated from differences in spatial extent of sample collection, for between-regions F ST comparison we used only CH, BO, SA, MU and HO populations from western region, which are spatially distributed similarly to those from eastern region (ODO, ODG, ODD and ODP). Due to low juvenile dispersal (Titov 2003), spotted suslik can form cryptic population substructure, and hence, for the relatedness coefficient, we used the Pamilo’s (1985) correction for the increased relatedness due to structuring of population (Chapuisat et al. 1997). For all the parameters except allelic richness, the average was weighted by sample size. In order to test the hypothesis that genetic diversity is higher and the relatedness is lower in the populations from the habitat with higher levels of connectivity for observed and expected heterozygosity and allelic richness, we used a one-sided test, stating that these parameters are higher in the eastern populations and the relatedness coefficient is higher in western populations. For the inbreeding coefficient, we used a two-sided test as its value can be affected not only by breeding of relatives but also by cryptic population structure. To assess the significance of the tests a permutation scheme was used.
Population differentiation was estimated with pairwise and overall F ST (Weir and Cockerham 1984), along with their significance determined with a permutation test as implemented in FSTAT. Partitioning of the genetic diversity among regions, among populations within regions and within populations, was investigated with a hierarchical analysis of molecular variance AMOVA (Excoffier et al. 1992) in Arlequin 3.01 (Schneider et al. 2000), with pairwise F ST as the distance measure. To assess how the genetic variation is partitioned within each region we also performed AMOVA analysis for eastern and western populations separately. Bayesian assignment test of STRUCTURE 2.0 (Pritchard et al. 2000) was used to assign individuals to clusters based on multi-locus genotypes with no prior population information. The analyses were performed independently for the western and eastern regions. In order to determine the most probable number of genetically distinct populations (K) within our dataset, we ran a simulation for each K (2–12) for western populations and K (1–6) for eastern populations with 100,000 burn-in iterations and 100,000 data iterations. The most likely value of K is the one that maximizes the log-likelihood of obtaining the observed sample of multilocus genotypes (Pritchard et al. 2000). A model with correlated allele frequencies and admixture among populations was used for both regions. Individuals were assigned to respective populations based on percentage of membership. To test for isolation by distance, we performed a regression analysis of Slatkin’s (1995) linearized F ST transformation (F ST/1 − F ST), on the natural log of geographical distance (Rousset 1997) for populations from the western and eastern region separately. In the western region, analyses were performed with the exclusion of population SW, which is known to be established by translocation of individuals from a different geographic location. The significance of this relationship was determined with a Mantel test (Mantel 1967) (10,000 permutations) using FSTAT. To check if known reductions in population sizes of Polish spotted suslik populations, or possible reductions of other populations, resulted in a decline of intrapopulation genetic variability, we performed a test of homozygosity excess using a Bottleneck software (Piry et al. 1999). During a population bottleneck, allelic diversity is lost more rapidly than heterozygosity (Nei et al. 1975; Maruyama and Fuerst 1985). Thus, a method for detecting recently bottlenecked populations is to compare the Hardy–Wienberg expected heterozygosity in a sample with the heterozygosity expected for mutation drift equilibrium. This heterozygosity excess should be detectable for approximately 0.2–4 Ne generations, where Ne is the effective population size. We tested for a heterozygote excess with a one-tailed Wilcoxon sign-rank test. We performed the analysis under the two-phase mutation model TPM with 90% of one-step mutations and 10% of multistep mutations, which is thought to most closely follow microsatellite mutation (Di Renzo et al. 1994). Second, we calculated Garza and Willamson’s (2001) ‘M’ value, implemented in Arlequin 3.01, relating the mean number of alleles to the total range in allele size. This method allows to distinguish stronger or more ancient size reductions than Bottleneck software (Garza and Willamson’s 2001).
Results
All 11 microsatellite loci were polymorphic. The total number of alleles varied from 3 to 15 per locus and overall locus heterozygosity ranged from 0.37 to 0.69 with an average 0.54 (SD = 0.112). Estimates of F IS at each locus in each population showed significant HW departure when Bonferroni correction was applied only in two out of 154 comparisons (Table 2). Genetic diversity indices are summarized in Table 3. The tests of HWE showed significant homozygote excess in three western populations (WY, SW and HO) but also in three eastern populations (ODG, ODO and ODP). There was no evidence for linkage disequilibrium. Genetic diversity was lower in the western populations than in eastern ones. Average values of observed and expected heterozygosity, as well as allelic richness, were significantly higher in populations from the eastern region. There was no significant difference for the average relatedness and F IS coefficient between the two regions as reported in Table 4.
Patterns of genetic differentiation among populations differed between the regions. For western populations, the values of F IT, F ST and F IS were respectively 0.28, 0.20 and 0.10 while for eastern colonies, the same indexes were as high as 0.20, 0.03 and 0.10, thereby displaying a much weaker structure at the interpopulation level in this region. As a measure of genetic divergence among populations, we used pairwise F ST and assignment tests. All colony pairs in the western region showed significant and very high genetic differentiation expressed by high interpopulation F ST, ranging from 0.05 (HO–MU) to 0.35 (BO–WY), with an average 0.21 (SD = 0.07). In the eastern region, pairwise F ST values ranged from 0.01 (ODO-ODD) to 0.05 (ODG–ODD and ODG–ODP) and in four comparisons, the value obtained was significantly different from 0. A hierarchical AMOVA, which examines the partitioning of genetic variation among populations versus regions, indicated that regions accounted for 18.37% of the total variability, 12.81% of variation was partitioned between populations within regions and 68.63% within populations (Table 5). The variation on each level was highly significant (P < 0.0001). AMOVA performed in each region separately resulted in a partitioning 27% of variation among populations in the western region while variation at the same level in the eastern region, accounted for only 3.76% of total variability. The Bayesian assignment test of STRUCTURE was used to identify the number of population clusters and simultaneously assign individuals to each of the identified clusters. Obtained clustering likelihoods supported a model with eight clusters in the western region and two clusters in the eastern region. The identified groups and the mean individual assignment proportions (q mean) are given in Tables 6 and 7 for the western and eastern region respectively. For each individual, the proportion of assignment to each cluster was also estimated. The highest proportion of membership for western populations was found in clusters representing individuals originating from those populations and this is consistent with the extremely high population structure. Only individuals from populations PO and HU were grouped in a single cluster. In Polish populations from the western region, no individual was assigned as a migrant with a proportion higher than 0.9. In Ukrainian populations from this region, individuals originating from population MU were assembled in two different clusters, representing individuals from two neighbouring populations (SA and HO). This result suggests recent migrations between these three populations. Also, one individual originating from population CH was identified as a migrant to the MU population (q > 0.9). In the eastern region, although two clusters were detected, individual admixture proportions indicated that most individuals were admixed between clusters. This pattern suggests high proportion of migrants or their progeny in each population (Table 8).
The Mantel test of matrix congruence revealed a significant positive correlation between pairwise F ST and the geographical distance between colonies in the western region (r = 0.72, P < 0.0001; Fig. 2), while there was no correlation for the same coordinates in eastern region.

Correlation between transformed genetic distance and geographic distance. Populations from eastern region indicated with open triangles
The Bottleneck test, reflecting the evidence of the recent reductions of effective population sizes, indicated significant (P < 0.005) heterozygosity excess in six out of 10 western populations. We did not find significant heterozygosity excess in CH, BO, SA and MU populations from western region. The M ratio higher than 0.8 indicates that population bottleneck was found in two western populations (TY; M = 0.82 and SW; M = 0.84). In eastern region only ODP population showed significant heterozygosity excess and no population displayed M ratio higher than 0.8, i.e. the test failed to indicate the bottleneck events.
Discussion
We used microsatellite markers to investigate the genetic diversity, population genetic structure and gene flow among colonies of spotted suslik in two separate regions characterized by different levels of habitat connectivity. Due to the fact that spotted suslik populations from the western border of its range are highly endangered because of habitat loss, but still numerous and connected in the eastern part of the range, we had a rare opportunity to compare the patterns of genetic diversity and the levels of gene flow in populations of the same species in two different habitats and to assess the impact of habitat loss on the genetic structure of populations. We used genetic diversity measures to test the hypothesis that changes in land use led to a reduction of genetic diversity within western populations. As those populations are located at the end of the species range, we tried to distinguish between the effects of recent and historical factors shaping the population structure of these populations.
Diversity and divergence of studied populations
The values of population diversity indices were markedly lower in the western region (Table 4). The observed heterozygosities of western populations are characteristic for endangered populations (Frankham et al. 2002; Hazlitt et al. 2006). Five out of ten populations from this region are fixed in at least one locus and the HU population is fixed in six out of 11 loci studied, while no monomorphic loci were detected in the eastern region. We also identified significantly lower, in comparison to the eastern region, values of allelic richness. This parameter is the most valuable indicator of the genetic condition of the population, as it reflects population ability to adapt to ecological perturbations (Lowe et al. 2003).
Microsatellite analysis revealed, not surprisingly, that spotted suslik from two different regions form two genetically different groups. All the analyses of inter population diversity proved very different levels of structuring in the two regions. Extremely strong divergence over short distances between populations in the western region contrasts with the relative genetic uniformity of populations in the eastern region. Although some degree of genetic differentiation between spotted suslik populations is expected because of its small dispersal ability, structure level in the western region is representative of strongly isolated populations. The values of population differentiation we obtained are comparable with those observed in the island populations of tuatara Sphenodon spp. (McAvoy et al. 2007) or Komodo dragon (Ciofi and Bruford 1999).
A number of different processes may potentially explain the patterns of microsatellite diversity and divergence that we observed in both regions. Lowered values of genetic diversity parameters in western populations could be explained by higher habitat fragmentation and extinction of many local populations over the last 50 years. It is also documented that all Polish populations of spotted suslik experienced recent population bottlenecks due to habitat loss (Męczyński 1991).
On the other hand, the effects of anthropogenic fragmentation in Polish populations of spotted suslik may be obscured by their location at the edge of the species range. Compared with populations from the central part of a species range, peripheral populations tend to be characterized by low density, high temporal variation and low effective population size (Gyllenberg and Hanski 1992; Schwartz et al. 2003; Vucetich and Waite 2003). Since all of these processes have contributed to current patterns of genetic diversity, the genetic signatures of these events could theoretically overwhelm population genetic changes, due to very recent anthropogenic fragmentation. Although Polish populations indeed are located at the edge of the species range, in our opinion the recent habitat deterioration was an important factor shaping the contemporary population structure in the western region. First, distribution of spotted suslik in Poland reported from the first surveys made by Surdacki (1963), before habitat destruction started, indicate that populations formed a system of abundant and well connected local populations. Second, despite strong divergence in the western region, we observed a significant correlation between geographic and genetic distance, indicating the existence of isolation by distance pattern. The lack of private alleles and very low private allelic richness in western populations also support recent interconnectivity between the populations in the western region (Slatkin 1985).
The results of traditional interpopulation differentiation analyses, such as pairwise F ST and AMOVA, are also supported by Bayesian clustering. Assignment tests revealed much higher levels of admixture in eastern populations, suggesting considerable intensity of migration between them. In contrast, assignment of individuals to clusters in the western region almost perfectly matched their populations of origin. Only in case of populations PO and WY, despite high pairwise F ST, individuals were placed in common cluster. This again could indicate gene flow between them in the recent past. Interestingly, individuals from the MU population from the western region, located in the Ukraine, were assigned to two different clusters characteristic for two neighbouring populations. This might result from the recent colonization of this population from adjacent populations or strong, directional gene flow between them.
Although we detected strong differences in population structure between regions we are aware that to some degree, this can result from larger spatial extent of sample collection in western region. Larger between-populations distances in this region could be the explanation of stronger population structure detected. Hence, for comparison of pairwise F ST values between two regions we excluded all remote western populations and used for comparison only populations BO, SA, MU, HO and CH, which represent spatial span similar to this from eastern region. We proved that even if only mentioned populations are used the difference in mean pairwise F ST is still significantly higher in the western region. Although there was no correlation between geographic and genetic distance in the eastern region, all pairwise F ST values were lower than for western region despite similar geographic scales for mentioned populations (Fig. 2). What is more, the differences in sampling extent are forced by present day situation in western region. Populations we sampled their are the only extant ones and large distances between them are caused by extinction of neighbouring colonies. On the other hand, distances between eastern populations represent spatial extent similar to this observed in western region before habitat destruction started.
We detected significant excess of homozygotes in a few populations in both regions. Although we are aware of possible occurrence of null alleles in microsatellite amplifications, we can rather rule out this phenomenon in our study. First, none of the loci have shown consequent deviation from Hardy–Weinberg equilibrium in the populations studied (Table 2). Furthermore, heterozygosity excess was detected in some loci and populations, however the deviation was statistically significant only in two out of 154 comparisons. Second, even in the populations with the most pronounced deficiency of heterozygotes, we obtained good quality PCR amplification for all samples in all loci. The most likely explanation of homozygosity excess is the mating between related individuals, which can be caused by various factors. Spotted suslik exhibit weak juvenile dispersal (Titov 2003) which may lead to creation of strong within population substructure. Although we were aware of these obstacles and performed animal sampling with special care, we cannot rule out completely the possibility of sampling some related individuals due to very small population sizes in some cases.
The extent to which genetic variability is lost in a population undergoing a demographic bottleneck is dependent on a number of factors: the scale and pattern of the loss of habitat, duration of the bottleneck, population numbers and the presence of fine-scale genetic structure (Frankel and Soulé 1981). Over longer periods, the combined effects of isolation and reduced migration would further influence this genetic variability.
The detection of genetic bottlenecks based on heterozygosity excess that detect the most recent bottlenecks is, in general, in accordance with the demographic bottlenecks documented in Polish populations (Męczyński 1991; Męczyński et al. 2001). It is known that all Polish populations experienced demographic bottlenecks and we detected significant heterozygote excess in five out of six of them. Two of these colonies also show an M ratio characteristic for a severe bottleneck, which is probably caused by a severe population reduction (Męczyński 1991; Męczyński et al. 2001). Interestingly, the only Polish population that did not exhibit any signs of bottleneck is population CH, located only a short distance from the Ukrainian ones. Together with the finding of possible migrants of Ukrainian origin in this population, we can assume that the observed reduction in size did not leave a footprint in the genetic data due to migrations from neighbouring populations. With the means of both tests, we detected significant size reduction in the SW population, despite its current number exceeding 11,000 individuals. This result can be easily explained by the ‘founder effect’ that surely took place when the population was established. The SW population was unexpectedly found in a grassy airport, further west outside the former spotted suslik range at the beginning of the 1990s. Thanks to perfect habitat conditions and the absence of natural predators, it grew in numbers very rapidly. Although we have no data on the historical demography of Ukrainian populations from both regions, the genetic bottleneck detected in four of them (two from the western region and two from the eastern), accompanied by relatively high genetic variation could probably be attributed to cyclic population size fluctuations or stochastic events, which is typical for many rodents. In population of Arvicola terestris, significant heterozygote excess was detected at the end of a low-density phase (Berthier et al. 2006).
Conservation guidelines
Conservation efforts to rescue extant spotted suslik populations were undertaken a few years ago and they comprise of restoring appropriate habitat through grass mowing, removal of higher vegetation and the tending of cattle in the places occupied by susliks. These practices are indispensable for the short-term preservation of endangered populations, but also in order to enable the long-term survival of spotted suslik in the western part of its range, genetic management should be undertaken. A few Polish populations, such as WY or HU are now extremely small and do not grow in numbers despite intensive habitat restoration efforts. The results reported here indicate that those populations also exhibit extremely low levels of genetic diversity. We cannot exclude the possibility that it is inbreeding depression that hampers the recovery of these populations; thus the exchange of individuals between the least numerous populations should be undertaken to increase the chances of the populations’ survival.
Within the active protection programme, the establishment of new populations in specially prepared reserves is also planned. This programme assumes utilizing individuals from the only large, self-sustaining population in Poland; namely population SW. However, as both our results and traditional subspecies classification based on morphological features do not indicate any obstacles for translocation of individuals between populations within the western area, this action should be carefully monitored. According to our findings, the SW population have recently undergone an extremely strong demographic bottleneck. Therefore, to maximize the chance of a successful reintroduction of genetic variation in the newly established populations, interbreeding with individuals from other Polish or western Ukrainian populations is recommended.
Future conservation activities should focus on the improvement of connectivity between existing populations, by restoring dispersal corridors with the added possibility of establishing stepping stone populations. Since Poland entered the European Union in 2004, this goal could be now partially achieved by modifying the EU’s agricultural programmes with more subsidiaries paid for maintaining pastures.
References
Amos W, Balmford A (2001) When does conservation genetics matter? Heredity 87:257–265
Barnett R, Yamaguchi N, Barnes I, Cooper A (2006) Lost populations and preserving genetic diversity in the lion Panthera leo: implications for its ex situ conservation. Conserv Genet 7:507–514
Berthier K, Charbonnel M, Galan M, Chaval Y, Cosson J-F (2006) Migration and recovery of the genetic diversity during the increasing density phase in cyclic vole populations. Mol Ecol 15:2665–2676
Chapuisat M, Goudet J, Keller L (1997) Microsatellites reveal high population viscosity and limited dispersal in ant Formica paralugubris. Evolution 51:475–482
Ciofi C, Bruford MW (1999) Genetic structure and gene flow among Komodo dragon populations inferred by microsatellite loci analysis. Mol Ecol 8:17–30
Di Renzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB (1994) Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA 91:3166–3170
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from genetic distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Frankel OH, Soule ME (1981) Conservation and evolution. Cambridge University Press, New York, New York
Frankham R, Ballou JD, Briscoe DA (2002) Conservation genetics. Cambridge University Press, Cambridge
Garner A, Rachlow JL, Waits LP (2005) Genetic diversity and population divergence in fragmented habitats: conservation of Idaho ground squirrels. Conserv Genet 6:759–774
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Głowaciński Z, Męczyński S (2001) Suseł perełkowany Spermophilus suslicus. In: Głowaciński (ed) Polish red data book of animals (Vertebrates). Państwowe wydawnictwa leśne i rolnicze. Warszawa, Poland
Gondek A, Verduijn M, Wolff K (2006) Polymorphic microsatellite markers for endangered spotted suslik, Spermophilus suslicus. Mol Ecol Notes 6:359–361
Gondek A (2004) Sytuacja susła perełkowanego w Polsce—zagrożenia i program ochrony. Biuletyn Monitoringu Przyrody 1/2004:21–27
Goudet J (2002) FSTAT (v. 1.2): a computer program to calculate F-statistics. J Heredity 86:485–486
Gyllenberg M, Hanski I (1992) Single-species metapopulation dynamics: a structured model. Theor Pop Biol 42:35–61
Hanslik S, Kruckenhauser L (2000) Microsatellite loci for two European sciurid species (Marmota marmota, Spermophilus citellus). Mol Ecol 9:2163–2165
Hazlitt SL, Goldizen AW, Eldridge MDB (2006) Significant patterns of population genetic structure and limited gene flow in a threatened macropodid marsupial despite continuous habitat in southeast Queensland, Australia. Conserv Genet 7:675–689
Hirota T, Hirohata T, Hiroshi M, Toshiyuki S, Yoshiaki O (2004) Population structure of the large Japanese field mouse, Apodemus speciosus (Rodentia: Muridae), in suburban landscape, based on mitochondrial D-loop sequences. Mol Ecol 13:3275–3282
Honnay LO Adriaens D, Coart E, Jacquemyn H, Roldan-Ruiz I (2007) Genetic diversity within and between remnant populations of the endangered calcareous grassland plant Globularia bisnagarica. Conserv Genet 8:293–303
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Lobkov VA (1999) Spotted suslik of the north-western region by the Black Sea: biology and regulation of it’s population. AstroPrint, Odessa, Ukraine
Lowe AJ, Jourde B, Breyne P, Colpaert N, Navarro C, Wilson J, Cavers S (2003) Fine-scale genetic structure and gene flow within Costa Rican populations of mahogany (Swietenia macrophylla). Heredity 90:268–275
MacAvoy ES, McGibbon LM, Sainsbury JP, Lawrence H, Wilson CA, Daugherty CH, Chambers GK (2007) Genetic variation in island populations of tuatara (Sphenodon spp) inferred from microsatellite markers. Conserv Genet 8:305–318
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Maruyama T, Fuerst PA (1985) Population bottlenecks and non-equilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics 111:675–689
May B, Gavin TA, Sherman PW, Korves TM (1997) Characterization of microsatellite loci in the northern Idaho ground squirrel Spermophilus brunneus brunneus. Mol Ecol 6:399–400
Męczyński S (1991) Występowanie susła perełkowanego Spermophilus suslicus w Polsce i koncepcje jego ochrony. Ochrona przyrody 48:207–237
Męczyński S, Grądziel T, Styka R, Duda P, Próchnicki K (2001) Inwentaryzacja stanowisk susła perełkowanego na Zamojszczyźnie w roku 2001. Spotted suslik monitoring evaluation- a raport for Lublin Voivodeship Nature Conservator, Lublin, Poland
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Nei M 1978: Estimation of average heterozygosity and genetic distance from a number of individuals. Genetics 89:538–590
Pamilo P (1985) Effect of inbreeding on genetic relatedness. Hereditas 103:195–200
Piry S, Luikart G, Cornuet J-M (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Heredity 95:536–539
Pritchard JK, Stevens M, Donnely P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 42:258–275
Ramirez O, Altet L, Ensen C, Vila C, Sanchez A, Ruiz A (2006) Genetic assessment of the Iberian wolf Canis lupus signatus captive breeding program. Conserv Genet 7:861–878
Rosenbaum PA, Robertson JM, Zamudio KR (2007) Unexpectedly low genetic divergences among populations of the threatened bog turtle (Glyptemys muhlenbergii). Conserv Genet 8:331–342
Rousset F, (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Schneider S, Roessli D, Excoffier L (2000): ARLEQUIN ver. 2.000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland
Schwartz MK, Mills LS, Ortega Y, Ruggiero LF, Allendorf FW (2003) Landscape location affects genetic variation of Canada lynx (Lynx canadensis). Mol Ecol 12:1807–1816
Slatkin M (1985) Rare alleles as indication of gene flow. Evolution 39:53–65
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Surdacki S (1963) Zmiany w rozmieszczeniu i liczebności Citellus suslicus na Lubelszczyźnie w okresie 1954–1961. Acta Theriol 7:79–89
Titov SV (2003) Juvenile dispersal in the colonies of Spermophilus major and S. suslicus ground squirrels. Russ J Ecol 34:255–260
Vucetich JA, Waite TA (2003) Spatial patterns of demography and genetic processes across the species range: null hypotheses for landscape conservation genetics. Conserv Genet 4:639–645
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Acknowledgements
This research was funded by the Polish State Committee for Scientific Research (grant no.: 3P04G00924) and the Polish Ministry of Science (grant no.: N30309031/2938).
We want to acknowledge Dr Vlodimir Lobkov from the Odessa National University, Ukraine for providing samples from the eastern region and to all the people who helped us to collect samples from the western region. We are also grateful to the Association of Zamość Natural Landscape Parks and the Lublin Voivodeship Nature Conservator for their support in our research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Biedrzycka, A., Konopiński, M.K. Genetic variability and the effect of habitat fragmentation in spotted suslik Spermophilus suslicus populations from two different regions. Conserv Genet 9, 1211–1221 (2008). https://doi.org/10.1007/s10592-007-9442-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-007-9442-8