
Abstract
We examined the genetic structure of snowy plovers (Charadrius alexandrinus) in North America, the Caribbean, and the west coast of South America to quantify variation within and among breeding areas and to test the validity of three previously recognized subspecies. Sequences (676 bp) from domains I and II of the mitochondrial control region were analyzed for 166 snowy plovers from 20 breeding areas. Variation was also examined at 10 microsatellite loci for 144 snowy plovers from 14 breeding areas. The mtDNA and microsatellite data provided strong evidence that the Puerto Rican breeding group is genetically divergent from sites in the continental U.S. (net sequence divergence = 0.38%; F ST for microsatellites = 0.190). Our data also revealed high levels of differentiation between sites from South America and North America (net sequence divergence = 0.81%; F ST for microsatellites = 0.253). In contrast, there was little genetic structure among breeding sites within the continental U.S. Our results suggest that snowy plovers in Florida should be considered part of C. a. nivosus (rather than part of C. a. tenuirostris, where they are currently placed), whereas snowy plovers from Puerto Rico should be considered part of C. a. tenuirostris. Snowy plovers in South America should remain a separate subspecies (C. a. occidentalis). Although U.S. Pacific and Gulf Coast breeding areas were not genetically distinct from other continental U.S. sites, demographic isolation, unique coastal habitats, and recent population declines suggest they warrant special concern.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The U.S. Endangered Species Act (ESA) allows listing of subspecies and other groupings below the species level, providing the U.S. Fish and Wildlife Service (USFWS) and the National Marine Fisheries Service (NMFS) with a means to target population units in greatest need of conservation (USFWS 1978). Currently, approximately one quarter of all ESA-listed taxa are subspecies, and for birds, 43% of listed taxa are subspecies (Haig et al. 2006). However, uncertainty regarding which criteria to use for delineating subspecies hampers subspecies listing and delisting decisions. Subspecies have been defined based on phenotypic (e.g., morphological, behavioral, and ecological) and genetic criteria (Mayr 1942; Amadon 1949; Futuyma 1998; Patten and Unitt 2002; Haig et al. 2004; Zink 2004). In birds, recent controversy over subspecies delineation has centered on conflicting morphological and genetic patterns (Zink 1989; Ball and Avise 1992; Zink et al. 2000; Zink 2004; Phillimore and Owens 2006). Part of this problem stems from reliance on a single locus with unique properties, mitochondrial DNA (mtDNA), for identifying subspecies (e.g., Zink 2004; reviewed by Ballard and Whitlock 2004). Patterns from a single locus may not accurately reflect genetic differences among populations because of random genetic effects. Moreover, divergent natural and/or sexual selection can maintain morphological differences among populations in the face of ongoing gene flow, depending on the strength of selection, migration rates, and the genetic architecture of the morphological traits in question (Haldane 1931; Lewontin and Krakauer 1975). Here we use several independent nuclear markers and mtDNA to test whether genetic structure corresponds with previously recognized subspecific boundaries in a threatened shorebird, the snowy plover (Charadrius alexandrinus).
Snowy plovers are wide-ranging, patchily distributed, and one of the most controversial threatened species in the U.S. Concerns largely stem from their association with beaches on the Pacific and Gulf coasts which are increasingly developed and used for recreation (Warriner et al. 1986; Chase and Gore 1989; Page et al. 1995; Lafferty 2001; Ruhlen et al. 2003). Snowy plovers are migratory and globally distributed, found in portions of North and South America and in Europe, Asia, and Africa (where they are called Kentish plovers; Cramp and Simmons 1983). Evidence suggests that the species is declining in Europe (Stroud et al. 2004) as well as North America (Page et al. 1995). Across the world, they inhabit coastal beaches, lagoons, salt-evaporation ponds, inland salt flats, and braided river channels (Wilson-Jacobs and Meslow 1984; Page et al. 1995). Some field studies suggest that snowy plovers have fairly high fidelity to breeding sites (Paton and Edwards 1996; Page 2004), although dispersal may be high enough to prevent substantial divergence in allele frequencies.
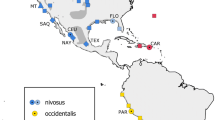
In the Western Hemisphere, three subspecies of snowy plovers have been recognized (Fig. 1; American Ornithologists’ Union 1957) based on differences in plumage coloration. Cuban snowy plovers (C. a. tenuirostris) breed on the Gulf Coast (east of Louisiana), the Bahamas, the north coast of the Yucatán Peninsula, the Greater and Lesser Antilles, and the islands off the north coast of Venezuela. Western snowy plovers (C. a. nivosus) breed elsewhere in the U.S. and Mexico. Finally, C. a. occidentalis occurs on the west coast of South America from southwestern Ecuador through western Chile. Blake (1977) questioned the validity of these subspecies and more recent accounts recognize only C. a. nivosus (including the former C. a. tenuirostris) and C. a. occidentalis (Hayman et al. 1986; Binford 1989; Sibley and Monroe 1990).

Map of snowy plover sampling sites included in current study. Sample site information is provided in Table 1. Distributions of currently recognized subspecies are shown in dark gray (western snowy plover, Charadrius alexandrinus nivosus), light gray (Cuban snowy plover, C. a. tenuirostris), and hatched pattern (C. a. occidentalis; modified from Cramp and Simmons 1983; Page et al. 1995)
The snowy plover population breeding on the Pacific Coast of the U.S. and Baja California is listed as a threatened distinct population segment (DPS; USFWS 1996) under the U.S. Endangered Species Act (ESA; USFWS 1993). Currently, distinctness of this DPS is under scrutiny. The southeastern U.S. population of the snowy plover is also under consideration for listing under the ESA.
Previous genetic analyses for a limited sampling of snowy plovers in North America and Puerto Rico found some support for recognition of two subspecies, but little support for a distinct Pacific Coast population of western snowy plovers (Gorman 2000). Support for subspecies recognition came from nuclear inter-simple sequence repeat (ISSR) loci (dominant markers), which identified a genetic discontinuity between breeding areas west versus east of the Rocky Mountains. Mitochondrial DNA sequence analysis did not identify this same break, but did find unique haplotypes in the Puerto Rico population. No detailed morphological analyses have been conducted for snowy plovers.
The goal of this study was to examine genetic structure in snowy plovers in the Western Hemisphere, including North America, the Caribbean, and South America, in more detail using additional sites and individuals, longer mtDNA sequences, and microsatellite loci. Because of high mutation rates, microsatellite loci tend to have a large number of independent alleles which provides high statistical power for detecting population structure (Kalinowski 2002). Thus, our goals were to determine: (1) how many snowy plover subspecies occur in the Western Hemisphere, and their geographic distributions; (2) whether Pacific Coast breeding aggregations are genetically distinct from Great Basin birds; and (3) whether there are genetic differences between southeastern U.S. snowy plovers and those further west.
Methods
Subspecies criteria
For the current analysis, we defined a subspecies as a subset of populations with consistent genetic differences from other subsets of populations at multiple independent loci, with genetic differences consisting of significant variation in microsatellite allele and mtDNA haplotype frequencies, the presence of unique alleles or haplotypes, and significant net sequence divergence. Such genetic differences would indicate low levels of gene flow among populations, and therefore greater potential to become full species than if populations were connected by high levels of gene flow (Mayr 1942, 1963; Futuyma 1998; Barrowclough and Vogler 2000; Coyne and Orr 2004). Thus, this criterion for defining subspecies is in line with the general idea of subspecies as incipient species (Darwin 1868; Frankham et al. 2002).
Although we define subspecies in terms of genetic criteria, we acknowledge that heritable phenotypic traits are equally valid for delimiting subspecies, and that both types of data should be used to define subspecies whenever possible. Moreover, we recognize that our definition is not perfect. For example, populations in the early stages of speciation may continue to have high levels of gene flow despite adaptive differences (Endler 1977). Conversely, populations with low levels of gene flow and high genetic differentiation may not have adaptive differences and may never become reproductively isolated (i.e., full species). Nonetheless, there are limitations in any subspecies definition and it is essential to explicitly define the criteria used to delineate subspecies prior to analysis (Haig et al. 2006).
Sample collection
Blood or tissue samples were collected from 166 individuals from 20 breeding areas from throughout the snowy plover breeding range in North America, the Caribbean, and South America (Table 1; Fig. 1). No known close relatives (e.g., parent/offspring, siblings, etc.) were included in the sampling and all samples were collected during the breeding season prior to immigration of birds from other sites. Six mountain plover (C. montanus) samples were also obtained to serve as an outgroup for phylogenetic analyses. A previous phylogenetic analysis of the genus Charadrius suggested that mountain plovers are a sister species of the clade which contains snowy plovers and red-capped plovers (C. ruficapillus; Joseph et al. 1999). Therefore, although snowy and mountain plovers are not sister taxa, they still have a relatively recent common ancestry.
Following protocols outlined by the American Ornithologists’ Union (Gaunt and Oring 1997), blood samples were collected from breeding populations in Puerto Rico, Florida, Kansas, Texas, Utah, eastern Oregon, and California (Table 1; Fig. 1). Once snowy plover nests were located, standard shorebird nest traps were used to catch adult birds as they returned to incubate (see Hill and Talent 1990). In order to minimize nest desertion, birds were not trapped unless at least two eggs had been laid. Birds were marked with a USFWS aluminum band to ensure that individuals were not resampled. Blood samples were taken from the brachial vein using a 27 gauge needle, collected in a 0.25 ml heparinized capillary tube, and transferred to a 2 ml cryogenic vial containing a buffer solution (100 mM Tris HCl, pH 8.0; 100 mM EDTA, pH 8.0; 10 mM NaCl; 0.5% SDS). Samples were stored at room temperature until they could be put into permanent storage at −80°C. In Washington, Oregon, California, Oklahoma, and Texas, heart, pectoralis, and embryonic tissue samples were collected from carcasses of individuals that died of natural causes, and membranes were collected from eggs in Colorado and Washington. Peruvian tissue samples were provided by the Louisiana State University Museum of Natural Science. Toe pad samples were acquired from specimens at the National Museum of Natural History from Bermuda, the Bahamas, Haiti, the Yucatán, and five additional individuals from Puerto Rico. The Yucatán specimen was collected in 1865, and all other NMNH specimens were collected between 1927 and 1936. All other samples were collected between 1995 and 2005.
Mitochondrial DNA methods
DNA was obtained from samples using standard phenol/chloroform extractions as previously described (Haig et al. 2004). All museum samples were extracted under clean conditions in our ancient DNA room separate from PCR products, and negative controls were run with all PCRs to ensure no contamination. An approximately 1.2 kb fragment of the mitochondrial control region was obtained using PCR primers L16525 and TS778 (Wenink et al. 1994; Sorenson et al. 1999). Amplifications were performed using a PTC 100 thermal cycler (MJ Research, Waltham, Massachusetts, USA). A total reaction volume of 20 μl was used with the following concentrations: 10 mM Tris–HCl at pH 8.3; 50 mM KCl; 0.001% gelatin; 3.5 mM MgCl2; 100 μM for each of the dNTPs; 0.2 μM of each primer; 100 ng of template; and 1.5 U AmpliTaq Gold Polymerase (Perkin Elmer, Wellesly, Massachusetts, USA). The following parameters were used for amplifications: 12 min denaturation at 93°C, followed by 35 cycles of 30 s at 93°C, annealing at 50°C for 30 s, and elongation at 72°C for 1 min. A final 10 min period of elongation at 72°C followed the last cycle. Successful PCR reactions were cleaned and concentrated by centrifugation dialysis using Microcon 30,000 MW cutoff filters (Amicon Bioseparations of Millipore Corp., Beverly, Massachusetts, USA). Due to a problematic poly C 5′ region in the control region, internal primers SNPL90 (SATCATGACACGTCACATGC) and SNPL 200 (CCCTAYCACATATCACCCA) were designed to generate bidirectional DNA sequence in domains I and II of the control region. Moreover, because of difficulty amplifying the 1.2 kb fragment from museum specimens, a shorter fragment (∼700 bp) was amplified using the SNPL90 and TS778 primers. Sequences were generated using ABI Prism Big Dye Terminator Cycle Sequencing chemistry on an ABI 3100 capillary DNA automated sequencer (377 DNA Sequencer ABI Prism 377XL Collection Software) located in the Central Services Laboratory at Oregon State University. Ambiguities were resolved by comparing light and heavy-strand sequences or from overlap of different fragments. Use of avian blood as a DNA source could allow amplification of nuclear homologs. However, our amplification of a large fragment (1.2 kb) of the mitochondrial genome as well as bi-directional sequencing increased the likelihood that DNA sequences were mitochondrial in origin. Also, samples containing ambiguous sequence (i.e., double peaks) were excluded from further analyses. Finally, there was no relationship between tissue type and haplotypes which would be expected if amplification of blood samples were resulting in nuclear homologs.
Initial alignment of DNA sequences was completed in ClustalX (Thompson et al. 1997). Manual adjustments were then made in MacClade 4.08 OSX (Maddison and Maddison 2000) so as to minimize the number of changes required across sites. There were two sites at which all mountain plovers had an insertion (or, alternatively, all snowy plovers had a deletion). These two indels were removed from subsequent analyses. Only unique haplotypes were included in matrices used for phylogenetic analyses. Parsimony analyses were conducted in PAUP* 4.0b10 (Swofford 2000) using a heuristic search with 100 random addition-sequence replicates and TBR branch swapping. Nodal support was assessed through non-parametric bootstrap analysis using 1,000 bootstrap replicates with 10 random addition-sequence replicates per bootstrap replicate.
The most appropriate model of sequence evolution for the likelihood analysis was selected using Akaike’s information criterion (Akaike 1974) in Modeltest 3.7 (Posada and Crandall 1998). Likelihood analysis was then conducted in PAUP* 4.0b10 using successive iterations with starting parameters based on estimates from the previous tree, a method shown to perform well (Sullivan et al. 2005). Parameters for the first iteration were estimated from the most-parsimonious tree with the best likelihood score. Iterations were continued until successive searches yielded identical trees, likelihood scores, and model parameters. Two replicate Bayesian analyses were conducted with MrBayes 3.04b (Huelsenbeck and Ronquist 2001). In each replicate, four Markov chains were used and the chain was sampled every 100 generations. Markov chains were run for five million generations. Program TCS was also used to construct a haplotype network from control region haplotypes using statistical parsimony (Clement et al. 2000).
We used all 166 snowy plover sequences for population genetic analyses, but did not use outgroup sequences (mountain plovers). All mtDNA population genetic analyses were performed using Arlequin version 3.01 (Excoffier et al. 2005). Genetic variation within sites was estimated using a variety of diversity statistics, including haplotype diversity (h), number of polymorphic sites (s), and nucleotide diversity (π n ). Tajima’s D was also used to test for population bottlenecks and population expansion (Tajima 1989). Corrected average percent sequence divergence, equivalent to net sequence divergence, was also calculated in Arlequin. Genetic divergence among sites was estimated using F-statistics and analysis of molecular variance (AMOVA; Excoffier et al. 1992) in Arlequin version 3.01 which takes into account the number of mutations between haplotypes. One thousand random permutations were used to test the probability of observing pairwise ΦST values as large as or larger than those observed by chance. Pairwise Φ ST values were calculated among all individual sites (with samples size of four or more individuals) and between North America (including Caribbean sites) versus South America, western versus Cuban snowy plovers, Pacific Coast versus Great Basin, Florida versus the rest of the continental U.S., and Puerto Rico versus the rest of North America. The α value for each test was determined using a sequential Bonferroni adjustment (Rice 1989). For the AMOVA analysis, we used four a priori groupings (North America versus South America; western versus Cuban snowy plovers; Pacific Coast versus Great Basin; and Florida versus the rest of the continental U.S.). One thousand random permutations were also used to test the significance of variance components. As only two birds were sampled in southern Florida, these birds were grouped with birds from the next closest sites in northern Florida for analyses. Similarly, as only one bird was available from the most northerly site in Peru and one bird was available from the most southerly site in Peru, we grouped all Peruvian samples together.
DNA sequences from this analysis have been deposited in GenBank under accession numbers (EF215532–EF215703).
Microsatellite methods
Microsatellite primer sequences were obtained from a number of sources (Table 2). Initially, 53 primers pairs previously isolated from eight different species were tested for cross-species amplification. From these, two (RBG-29, HrU2) (Primmer et al. 1995; Given et al. 2002) were polymorphic and gave reproducible results. RBG-29 was later excluded from analyses because of the presence of non-microsatellite insertion–deletions. Primers CALEX-10, 19, 32, 35, 39 (developed for Kentish plovers) were obtained from Küpper et al. (2007).
Microsatellite loci C201, C203, C204, and C205 were obtained from ISSR-suppression-PCR clone libraries (Lian et al. 2001). Briefly, PCR fragments obtained from amplifications with primers (CA)8, (AC)7, (AG)7, (GA)7, (TC)7, (AT)7, (GT)7 were used for ligation into a PCR-Script Cam cloning vector (Stratagene) and were subsequently subcloned and positive clones sequenced. From these libraries, 19 microsatellite sequences were chosen to design nested PCR primers (IP1 and IP2) for the determination of unknown flanking regions. Adaptor-ligated, restricted DNA libaries were constructed with AluI or HaeIII digested DNA fragments ligated to a suppression PCR adaptor consisting of a 48-mer single-stranded oligonucleotide (5′-GTAATACGACTCACTATAGGGCACGCGTGGTCGACGGCCCGGGCTGGT-3′) and the complementary 8-mer 3′-end amino capped oligo (5′-ACCAGCCC-NH2-3′). Nested PCR amplifications were performed using primer pairs AP1/IP1 and AP2/IP2 and 13 PCR products forming a single band were sequenced. Primers (IP3) were designed for the newly defined flanking sequence and characteristics of the isolated microsatellites were investigated. Four of these sequences provided reproducible polymorphic loci and were used for further analysis.
For screening samples, PCR amplifications were performed in 10 μl reactions using the same conditions as described previously for mitochondrial amplifications. Annealing temperatures for primers are shown in Table 2. Amplification products were analyzed on an ABI 3100 capillary DNA automated sequencer located in the Central Services Laboratory at Oregon State University. ABI Genescan® analysis software was used to determine the size of fragments based on internal lane standard GeneScan 500 [Rox]. ABI Genotyper® software was used to score alleles accurately. Of the 166 snowy plover samples available, our 10 microsatellite loci amplified successfully in 144 individuals collected between 1995 and 2005. Loci did not consistently amplify for museum samples collected between 1865 and 1936 or for individuals collected recently from Santa Barbara (California) or Colorado, and therefore were not included in the analysis.
Allele frequencies, exact probabilities for Hardy–Weinberg proportions, and exact probabilities for genotypic disequilibrium were calculated using Genepop version 3.4 (Raymond and Rousset 1995). Expected heterozygosities were calculated in Microsatellite Analyzer (Dieringer and Schlötterer 2003) and allelic richness was calculated in FSTAT version 2.9.3.2 (Goudet 2001). Allelic richness is an index of the number of alleles corrected for sample size using rarefaction (El Mousadik and Petit 1996). We tested for an excess of heterozygotes beyond that expected in a population at mutation-drift equilibrium, indicative of population bottlenecks, for four groupings of sites with at least 10 individuals (Pacific Coast = WA, ORC, HUMB, MONT, and SCA in Fig. 1; Great Basin = ORSL and UT; Midwest = KS and OK; and the Gulf Coast = TX, LA, and FL) using program BOTTLENECK (Cornuet and Luikart 1996).
Genetic divergence among sites was assessed with F-statistics and AMOVA using Genepop version 3.4 (for calculating overall F ST) and Arlequin version 3.01 (for calculating pairwise F ST values and AMOVA), as described above for mtDNA. We also conducted principle components analysis (PCA) using Minitab version 14 as a method of assessing overall genetic similarity among populations. The largest allele at each locus was omitted to account for non-independence of alleles within each locus. We then plotted PC2 versus PC1 and PC3 versus PC2 to estimate genetic divergence as the relative linear distance between points representing each population. Isolation-by-distance among individuals was tested using Mantel tests as implemented in Alleles In Space (AIS; Miller 2005). The distance measure used in AIS is analogous to that used by Nei et al. (1983) for population frequency data, but is instead applied to pairs of individuals rather than pairs of populations. Therefore, no a priori assumptions regarding the geographic delineation of populations are necessary.
We also used a Bayesian clustering approach implemented in program STRUCTURE version 2.1 (Pritchard et al. 2000) to estimate the number of populations (K) in a sample and to assign individuals to one or more of these populations (k). This approach groups populations by minimizing deviations from Hardy–Weinberg equilibrium and linkage equilibrium between loci within populations. We used the admixture model which assumes gene flow among populations. The admixture model assigns a proportion of each individual’s genome to each population (q k ). We calculated the probability that there are from K = 1 to 14 populations (the total number of sites for which microsatellite data were available). We ran five independent simulations for each K, used a burn-in length of 50,000 and a run length of 106, and assumed correlated allele frequencies.
Finally, we estimated gene flow (Nm) among populations and theta (Θ = 4Nμ) within populations using the coalescent approach implemented in program MIGRATE (Beerli and Felsenstein 2001). For this analysis, we grouped sites into six populations (the same four groupings shown above for the population bottleneck analysis plus Puerto Rico and Peru). For each run of MIGRATE, we used 10 short chains of 10,000 sampled and 500 recorded trees, followed by three long chains of 100,000 sampled and 5,000 recorded trees. Four-chain heating was used with temperatures set to 1, 1.5, 3, and 6 in order to improve sampling of tree space. We conducted this analysis four times with different random number seeds. Initial estimates of theta and 4Nm were generated from F ST values for the first run, and the maximum-likelihood estimates from the previous run were used as the initial estimates of these parameters for the subsequent three runs to confirm that final chains converged on the same estimates (as determined by overlapping 95% confidence intervals). MIGRATE assumes migration-drift equilibrium. Estimates from the last run are reported (Table 6).
All sample, mtDNA, and microsatellite data are available from the corresponding author upon request.
Results
Mitochondrial DNA variation
In the final alignment, 99 out of 676 characters were variable, and 74 were parsimony informative. There were a total of 57 unique haplotypes for the 166 snowy plover individuals sequenced and five unique mountain plover haplotypes out of six individuals sequenced.
Maximum-parsimony analysis generated 18,899 most-parsimonious trees of 143 steps (CI = 0.727, RI = 0.902). The best model of sequence evolution chosen by Modeltest 3.7 was a TrN + Γ + I model (Tamura and Nei 1993 model with two transition classes and one transversion class, among-site rate heterogeneity, and invariant sites). The maximum likelihood analysis only required two iterations to reach convergence of tree topologies and branch lengths (Fig. 2). The negative log-likelihood score of the two final trees was 1,744.93 (estimated base frequencies: A: 0.2439, C: 0.2821, G: 0.1607, T: 0.3133; rate matrix: A–C: 1.0, A–G: 9.076, A–T: 1.0, C–G: 1.0, C–T: 17.809, G–T: 1.0; shape parameter for gamma distribution: 0.6459; proportion of invariant sites: 0.6050).
For the Bayesian phylogenetic analysis, plots of model parameters and likelihood versus generation number indicated that stationarity had been reached by generation 100,000. Moreover, bipartition posterior estimates obtained from all samples after removal of a conservative burn-in period of 5,000 (equivalent to 500,000 generations) appeared to converge in pairwise comparisons between the two runs (using the comparetree command in MrBayes). Therefore, the last 4.5 million generations (equivalent to 45,000 sampled trees) of each of the two runs were combined to yield 90,000 trees for the final Bayesian posterior probabilities (bpp). The 50% majority rule consensus tree of all Bayesian trees had 12 nodes with greater than 50% support, all of which were also observed in the maximum likelihood tree. Bayesian posterior probabilities for these nodes are shown on the maximum likelihood tree (Fig. 2).

Maximum likelihood topology from analysis of mtDNA control region for snowy plovers. Haplotype numbers are shown in bold and site codes (which correspond to codes used in Table 1 and Fig. 1) are shown in parentheses. Numbers shown at nodes are Bayesian posterior probabilities from 90,000 sampled trees. Outgroup taxa are not shown
In all analyses (parsimony, likelihood, and Bayesian), few clades were well supported due to low phylogenetic signal in the mtDNA control region. Most clades also had little association with geography. For example, the clade containing haplotypes 1–15 includes 14 of the 20 sites analyzed in this study, ranging from the Pacific Coast of the U.S. to Puerto Rico. Also, many haplotypes (e.g., haplotypes 14, 31, and 45) were geographically widespread, spanning the entire North American continent. However, there were two clades that were geographically restricted. First, the clade containing haplotypes 6–9 was restricted to Puerto Rico, and was very well supported (100% bpp). Additionally, the clade containing haplotypes 52–57 was restricted to Peru and was also fairly well supported (79% bpp).
The haplotype network generated in TCS also reveals that many haplotypes were shared by different currently recognized subspecies (Fig. 3). Specifically, haplotypes 14, 27, 31, 43, and 45 were shared by both western and Cuban snowy plovers. However, the haplotype network, like the maximum likelihood phylogeny (Fig. 2), shows a distinct clade for haplotypes 6–9 from Puerto Rico and another distinct clade for haplotypes 52–57 from Peru.

Statistical 95% parsimony network generated by TCS based on mtDNA control region haplotypes for snowy plovers. Circle sizes are proportional to the number of individuals sharing the haplotype. Haplotype numbers are in circles. Shades refer to the proportion of samples that came from a traditional subspecies designation: western snowy plover (Charadrius alexandrinus nivosus) haplotypes are in black; Cuban snowy plover (C. a. tenuirostris) in gray; and South American snowy plover (C. a. occidentalis) in white. Dashes are inferred haplotypes
Corrected average percent sequence divergence (equivalent to net percent sequence divergence) between different groupings of snowy plovers is shown in Table 3. Net sequence divergence between recognized western and Cuban subspecies was low but statistically significant. Net sequence divergence between Pacific Coast and Great Basin birds and between Florida and other continental U.S. birds was low and not significant. In contrast, net sequence divergence was relatively high and statistically significant for the comparisons between North America versus South America, western snowy plovers versus South America, Cuban snowy plovers versus South America, and Puerto Rico versus the continental U.S. The mean number of pairwise differences between Puerto Rico and all North American and Caribbean sites was 6.8 (range: 0–14). Moreover, these substitutions were dispersed throughout the control region, suggesting that they were independent mutation events. The mean number of pairwise differences between Peru and North America was 9.9 (range: 1–17), and these substitutions were also dispersed throughout the control region.
Population genetic analyses of mtDNA sequences revealed substantial variation among sites at the level of within population genetic variation as measured by haplotype and nucleotide diversity (Table 1). Haplotype diversity was lowest in Louisiana and Bermuda and highest in Humboldt County (California), Colorado, Oklahoma, the Bahamas, Haiti, and Peru. Nucleotide diversity was also lowest in Louisiana and Bermuda, and highest in Colorado, Puerto Rico, and Peru. No Tajima’s D values were significant, providing no evidence for population bottlenecks or expansions.
Population genetic analyses also revealed a non-random distribution of sequence variation among populations, despite poor phylogenetic resolution and a general lack of association of clades with geography. Pairwise ΦST values ranged from negative values between many different pairs of sites up to 0.777 between Louisiana and Monterey Co., California (Table 4). Pairwise ΦST values between Puerto Rico and all other sites were large (mean: 0.378; range: 0.284–0.490), as were pairwise ΦST values between Peru and all other sites (mean: 0.609; range: 0.490–0.704). Eight pairwise comparisons (53%) between Puerto Rico and other sites were significant; 11 pairwise comparisons (73%) between Peru and other sites were significant. Pairwise ΦST values between North versus South America (ΦST = 0.603), western versus Cuban snowy plovers (ΦST = 0.126), Florida versus the rest of the continental U.S. (ΦST = 0.088), and Puerto Rico versus other North American sites (ΦST = 0.443) were all significant, but the pairwise Φ ST value between Pacific Coast versus Great Basin birds (Φ ST = −0.003) was not significant.
A priori groupings of snowy plover sites were poorly supported by AMOVA, except for grouping by continent (Table 5). Grouping of sites by continent (North versus South America) explained 58.39% of the variation in haplotype frequencies, although this grouping was only marginally significant due to the small sample size available from Peru (P = 0.052). Grouping of sites by currently recognized western versus Cuban snowy plovers was statistically significant (P = 0.004) because of much larger sample sizes in each group, but only explained 9.79% of the variation in haplotype frequencies. Grouping western sites into Pacific Coast versus Great Basin groups explained −3.00% of the variation (the negative value indicates that haplotype frequencies tended to be more similar among groups than they were within groups; P = 0.738). Grouping of sites into Florida versus the rest of the continental U.S. groups also only explained 6.39% of the variation (P = 0.203).
Microsatellite variation
Genotypic frequencies conformed to expected Hardy–Weinberg proportions. Only four of 120 tests for deviation from Hardy–Weinberg proportions were statistically significant at the α = 0.05 level which is less than the six tests expected to deviate by chance. No loci had a consistent excess of homozygotes as would be expected if there were null alleles. Moreover, no tests for linkage disequilibrium were significant at the α = 0.05 level, indicating all 10 loci were independent.
Overall levels of genetic variation within sites varied substantially among sites and loci. Average expected heterozygosity ranged from 0.249 in Puerto Rico to 0.539 at Monterey, California (Table 1). Allelic richness ranged from 1.70 in Puerto Rico to 2.49 in Texas (Table 1). The number of alleles per locus also varied substantially among loci, ranging from two alleles at C205 to 10 at C204 (Table 2). Of the three tests for heterozygote excess implemented in program BOTTLENECK (sign test, standardized differences test, and Wilcoxon sign rank test), only the Wilcoxon test was significant (P = 0.02) for the Pacific Coast population. Thus there was limited evidence for a population bottleneck in the Pacific Coast population, and no support for bottlenecks in the other three populations (Great Basin, Midwest, and Gulf Coast).
The overall F ST value among all sites was 0.069 and overall genetic differentiation was statistically significant (P < 0.001). Pairwise F ST values revealed that significant population structure was largely due to two sites, Puerto Rico and Peru, as observed with mtDNA haplotypes (Table 4). Pairwise F ST values between Puerto Rico and all other sites were large (mean: 0.256; range: 0.169–0.362), as were pairwise F ST values between Peru and all other sites (mean: 0.282; range: 0.209–0.362). Eleven pairwise F ST values (85%) for comparisons between Puerto Rico and other sites were significant; eight pairwise F ST values (62%) for comparisons between Peru and other sites were significant. Additionally, three pairwise F ST values (27%) between Florida and other sites in the continental U.S. were significant. Pairwise F ST values between North and South America (F ST = 0.253), western versus Cuban snowy plovers (F ST = 0.028), Florida versus the rest of the continental U.S. (F ST = 0.036), and Puerto Rico versus the continental U.S. (F ST = 0.190) were all significant, but the pairwise F ST value between Pacific Coast versus Great Basin birds (F ST = 0.002) was not significant.
There were two unique alleles at two different loci in Puerto Rico and four unique alleles at four different loci in Peru (Appendix Table 7). Moreover, one allele unique to Puerto Rico (allele 4 at C203) and three alleles unique to Peru (allele 5 at C203, allele 4 at CALEX-19, and allele 2 at CALEX-35) were found at high frequencies, ranging from 0.333 to 0.500.
Similar to mtDNA sequences, a priori groupings of snowy plover sites were poorly supported by AMOVA for microsatellite loci except for grouping by continents (Table 5). Grouping of sites by continent (North versus South America) explained 23.84% of the variation in allele frequencies, but was not significant (P = 0.075) due to the small sample size from Peru. Grouping of sites by subspecies (western versus Cuban snowy plovers) only explained 1.51% of the variation in allele frequencies (P = 0.166). Similarly, grouping western sites into Pacific Coast versus Great Basin groups explained −0.16% of the variation (P = 0.587). Grouping sites into Florida versus the rest of the continental U.S. also only explained 3.06% of the variation (P = 0.181).
Principle components analysis revealed high divergence between Peru, Puerto Rico, and all other sites in the continental U.S. (Fig. 4). Peru was highly separated from all other sites along the first principle component axis (PC1), which explained 28.3% of the variation in allele frequencies and had high negative loadings for the four alleles unique to Peru. Puerto Rico was primarily separated from other sites along the third principle component axis (PC3), which explained 12.7% of variation and had high positive loadings for two alleles unique to Puerto Rico, and to a lesser extent was separated from other sites along the second principle component axis (PC2) which explained 13.4% of the variation. Finally, Mantel tests revealed that a small, but significant proportion of the variation in genetic distance among individuals was explained by geographic distance (r = 0.363, P = 0.001; Fig. 5).


Genetic distance among individual snowy plovers at microsatellite loci versus geographic distance calculated in program AIS (r = 0.363, P = 0.001)
Program STRUCTURE clustered individuals into three groups with a posterior probability close to 1.0. Between 88% and 99% of the genomes of individuals from Puerto Rico and Peru were assigned to one group, whereas the genomes of individuals from throughout the U.S. were fairly evenly split between the other two groups. Thus the two continental U.S. groups identified by STRUCTURE were not associated with geography.
Gene flow (Nm) estimates from program MIGRATE were generally higher among continental U.S. populations (mean = 1.47; range = 0.54–3.76) than between U.S. populations, Puerto Rico, and Peru (mean = 0.16; range = 0.00–0.56; Table 6). The highest Nm estimates were from the Pacific Coast to other continental U.S. populations (mean = 3.14; range = 2.55–3.76) and the lowest Nm estimates were between Puerto Rico and Peru (mean = 0.07; range = 0.00–0.13). Theta estimates were higher in the continental U.S. (mean = 0.72; range = 0.58–0.78) than in Puerto Rico (0.30) or Peru (0.39; Table 6).
Discussion
Snowy plover subspecies
Our microsatellite and mtDNA data provide strong evidence that snowy plovers from Puerto Rico, the west coast of South America, and North America are genetically distinct from each other, but provide little evidence for population structure within the continental U.S. This supports current recognition of three subspecies of snowy plovers in the Western Hemisphere, but suggests that the geographic boundaries between western (Charadrius alexandrinus nivosus) and Cuban (C. a. tenuirostris) subspecies should be changed. Specifically, the border between western and Cuban snowy plover subspecies has traditionally been placed between Louisiana and Florida breeding populations (American Ornithologists’ Union 1957). Our data, however, suggest that the division between these subspecies should be between Puerto Rico and Florida. Under this recommendation, snowy plovers from the continental U.S. would be considered C. a. nivosus and birds from Puerto Rico would be part of C. a. tenuirostris. Additonal sampling and analyses of snowy plovers from the Caribbean, central Mexico, and the islands off the north coast of Venezuela will be necessary to determine the subspecies identity of these populations.
There are several lines of support for our subspecies delineations. First, all pairwise ΦST values for mtDNA haplotypes between Puerto Rico and continental U.S. sites and between South America and North America were large and most were statistically significant (Table 4). Second, six of the eight haplotypes from Puerto Rico were unique to the site (frequencies of haplotypes = 0.08–0.23) and all six of the haplotypes from Peru were unique to Peru (frequencies = 0.17). Third, net sequence divergence between individuals from Puerto Rico and the rest of the continental U.S. was fairly high (0.38%) and statistically significant, as was net sequence divergence between Peru and North America (0.81%; Table 3). However, sampling from other Caribbean islands was inadequate to determine their subspecies identity at this point.
The level of sequence divergence between snowy plovers from Puerto Rico and the continental U.S. and between the west coast of South America and North America is in the range of values observed among recognized avian subspecies for the mtDNA control region (0.49–2.90%; Fry and Zink 1998; Bhagabati et al. 2004). Moreover, Fry and Zink (1998) and Bhagabati et al. (2004) apparently used gross sequence divergence which does not remove within group sequence divergence and which is therefore expected to be higher than net sequence divergence as used here. Moreover, pairwise ΦST values between snowy plovers from Puerto Rico and snowy plovers from all other sites (mean = 0.378) and between South and North America (mean = 0.609) are within the range of values observed among avian subspecies at the mtDNA control region (0.036–0.950; Fry and Zink 1998; Valliantoes et al. 2002; Benedict et al. 2003; Eggert et al. 2004; Idaghdour et al. 2004; Pitra et al. 2004; Solorzano et al. 2004). These data demonstrate that mtDNA divergence between Puerto Rico and U.S. snowy plovers and between South and North America is well within the range seen in other well-established avian subspecies.
Our microsatellite data also provide strong evidence that Puerto Rican snowy plovers are genetically distinct from continental U.S. snowy plovers and that Peruvian snowy plovers are distinct from North American snowy plovers. First, as with mtDNA haplotypes, many pairwise F ST values between Puerto Rico and continental U.S. sites were significant at microsatellite loci and ranged from 0.169 to 0.317. Similarly, most pairwise F ST values between Peru and North American sites were significant at microsatellite loci and ranged from 0.209 to 0.362. These pairwise F ST values between snowy plovers from Puerto Rico and snowy plovers from the continental U.S. were also in the middle of the range of values observed among recognized avian subspecies at microsatellite loci of 0.023–0.571 (Chan and Arcese 2002; Eggert et al. 2004; Pitra et al. 2004; Jones et al. 2005). As with our mtDNA data, this shows that microsatellite variation between Puerto Rico and continental U.S. snowy plovers and between South and North America is within the range of differences seen among well-established avian subspecies. Additionally, gene flow (Nm) estimated with program MIGRATE was very low among the continental U.S., Puerto Rico, and Peru (Table 6). Finally, STRUCTURE identified Puerto Rico plus Peru as a group distinct from the continental U.S. STRUCTURE did not separate Puerto Rico and Peru into two different groups, but this is inconsistent with the high F ST values and low Nm estimates between these two populations, and is likely due to the relatively low power of STRUCTURE to identify populations (Waples and Gaggiotti 2006) and small sample sizes from these two sites.
It is important to note that the presence of two unique alleles in Puerto Rico and four unique alleles in Peru suggests that the high F ST values between Puerto Rico and continental U.S. sites and between Peru and North America are not simply due to sampling error stemming from the small sample sizes that were available from Puerto Rico and Peru. Detection of unique alleles at these sites despite small sample sizes strongly suggests that these alleles occur at reasonably high frequencies in these populations. Further, one of two unique alleles in Puerto Rico and three of four unique alleles in Peru were at high frequencies. If there were substantial gene flow between Puerto Rico and the continental U.S. or between Peru and North America, there would not be unique alleles at high frequency in these populations. Thus, detection of unique alleles despite limited sample sizes strongly suggests that snowy plovers from Puerto Rico are divergent from continental U.S. sites and that snowy plovers from Peru are divergent from North American sites at microsatellite loci as well as mtDNA.
In contrast, we found no genetic evidence for recognizing separate snowy plover subspecies within the continental U.S. First, no mtDNA clades were restricted to any sites in the western or eastern portion of the species’ continental U.S. range that might correspond with subspecies breaks (Figs. 2, 3). Second, there were no significant pairwise ΦST values between Florida and other sites in the continental U.S. for mtDNA haplotypes, and only a few significant pairwise F ST values between Florida and other sites in the continental U.S. for microsatellite loci (Table 4). Moreover, these F ST values were fairly low (pairwise F ST between Florida and other continental U.S. sites at microsatellite loci was 0.036), yet within the range of F ST values observed among populations of other avian species (range = 0.004–0.350; Caizergues et al. 2003; Johnson et al. 2003; Randi et al. 2003; Gay et al. 2004). This low F ST value indicates that Florida has historically been connected with other sites in the continental U.S. by fairly high levels of gene flow. Moreover, grouping samples by purported subspecies only explained a small proportion of the variance in mtDNA haplotype and microsatellite allele frequencies in the AMOVA analyses (Table 5). Gene flow estimates from program MIGRATE were also relatively high among populations in the continental U.S. (Table 6). Program STRUCTURE identified two groups in the continental U.S., but the genomes of individuals from across the mainland U.S. were evenly split between these two groups (i.e., there was no association between these two groups and geography). This pattern suggests that there is no population structure in the continental U.S. and that these sites should be lumped into a single group (Pritchard et al. 2000). These results for continental U.S. snowy plovers are concordant with a recent mtDNA analysis of mountain plovers in the U.S. western Great Plains and Colorado Plateau in which Oyler-McCance et al. (2005) also found little spatial genetic structure. Similarly, analysis of mtDNA and microsatellite variation among North American populations of least terns (Sterna antillarum) revealed low levels of population differentiation (Draheim 2006).
Gorman (2000) argued that the Rocky Mountains separate western and Cuban snowy plover subspecies based on genetic variation at ISSR loci, although her mtDNA data did not support this. In our study, we found no genetic evidence for such a break for mtDNA or microsatellite loci despite our larger sample sizes and the high statistical power of microsatellites for detecting population structure. However, we suggest that Gorman’s data are more consistent with gradual isolation by distance than with a distinct genetic break occurring across the Rocky Mountains. First, Gorman’s multivariate analysis of ISSR allele frequencies (Fig. 5 in Gorman 2000) indicates some overlap between populations east and west of the Rocky Mountains along axis 1 and complete overlap along axis 2. Second, she found highly significant isolation-by-distance at ISSR loci (Fig. 6 in Gorman 2000), suggesting that genetic differences on either side of the continent are due to geographic distance rather than subspecific breaks. Therefore, we argue that there is little support for two different subspecies of snowy plovers in the continental U.S. based on genetic data. Moreover, apparent lack of phenotypic differences among regions within the continental U.S. supports this (Blake 1977; Hayman et al. 1986; Binford 1989; Sibley and Monroe 1990).
Pacific Coast versus Great Basin snowy plovers
We found no evidence that Pacific Coast snowy plovers (Washington, Oregon Coast, Humboldt County, Monterey County, Santa Barbara County, and southern California; Fig. 1) are genetically distinct at neutral loci from Great Basin snowy plovers (Summer Lake and Abert Lake, Oregon, and Great Salt Lake, Utah). First, no mtDNA clades were restricted to Pacific Coast or to Great Basin sites (Fig. 2). Second, there were no significant pairwise Φ ST or F ST values between Pacific Coast sites and Great Basin sites for mtDNA or microsatellites, respectively (Table 4). Moreover, pairwise ΦST and F ST values between Pacific Coast and Great Basin snowy plovers for mtDNA and microsatellites, respectively, were still insignificant after lumping all Pacific Coast sites into one population and all Great Basin sites into another population. Therefore, lack of statistical significance between these two regions is not due to low statistical power. Additionally, none of the variance in mtDNA haplotype or microsatellite allele frequencies was explained by grouping samples into Pacific Coast and Great Basin groups in the AMOVA analysis (Table 5). Gene flow estimates between the Pacific Coast and the Great Basin were also the highest observed in this study (Table 6). Thus, all analyses indicate that there is no population substructure between Pacific Coast and Great Basin snowy plovers.
Lack of genetic differentiation is not unexpected within and between Pacific Coast and Great Basin breeding aggregations based on field studies documenting long-distance dispersal among sites (Stenzel et al. 1994; Page et al. 1995; Page 2004). While relatively high breeding site fidelity has been reported in these areas (Paton and Edwards 1996; Page 2004), within a single breeding season birds from the central California coast have been observed as far south as San Diego County (615 km) and inland at Summer and Abert Lakes, Oregon, in the western Great Basin (660 km; Stenzel et al. 1994). Between seasons, birds from central California have moved as far north as Washington (1,140 km). Additionally, natal and breeding dispersal has been observed from the California coast inland to Abert Lake (Page 2004). It is difficult to document the extent of this population exchange because no Great Basin sites are intensively monitored for banded birds.
We emphasize that only a few dispersers per generation are necessary to homogenize gene pools between breeding habitats (Wright 1931; Slatkin 1985, 1987; Mills and Allendorf 1996). Therefore, lack of significant genetic differences between Pacific Coast and Great Basin populations of snowy plovers does not necessarily mean that there is a high level of movement between these areas from a demographic standpoint. Although a few individuals are sufficient to prevent genetic differentiation among populations, a few individuals are not sufficient to maintain demographic connectivity. For example, if a Pacific Coast population of snowy plovers went extinct, a few immigrants from the Great Basin may not be sufficient to recolonize the empty habitat patch. For example, currently empty patches of Pacific Coast habitat are not being recolonized so there is no reason to suspect it will happen in the future when habitat is more fragmented.
Florida versus other continental U.S. sites
We also found little evidence that snowy plovers from the southeastern U.S. (Florida) are genetically distinct from snowy plovers in the rest of the continental U.S. First, no mtDNA clades were restricted to Florida (Fig. 2). Second, there were few significant pairwise ΦST and F ST values between sites in Florida and other continental U.S. sites for mtDNA and microsatellites, respectively, and low mean pairwise ΦST and F ST values indicated fairly high levels of gene flow (Table 4). Pairwise ΦST and F ST values between Florida and all other sites grouped into a single population were statistically significant for mtDNA and microsatellites, respectively, due to increased statistical power, but the fairly low values for this comparison (ΦST for mtDNA: 0.088; F ST for microsatellites: 0.036) suggest substantial levels of gene flow. Additionally, only a small proportion of the variance in mtDNA haplotype and microsatellite allele frequencies was explained by grouping sites into Florida versus the rest of the continental U.S. in the AMOVA analysis (Table 5). The Gulf Coast was also connected to other continental U.S. populations by fairly high levels of gene flow (Table 6). All analyses, therefore, indicated that there is minimal population substructure between snowy plovers in Florida versus the rest of the continental U.S. We are unaware of any data on snowy plover dispersal for the southeastern U.S., but based on the observations of long-distance dispersal among snowy plover breeding areas in the western U.S. cited above, lack of genetic differentiation is also not unexpected between Florida and other continental U.S. sites. Once again, we emphasize that lack of significant genetic differences between snowy plovers from Florida and elsewhere in the continental U.S. does not necessarily indicate a high level of movement between these areas from a demographic standpoint, nor absence of adaptive genetic variation.
Conservation implications
Our data provide strong evidence that snowy plovers from Puerto Rico are genetically distinct from snowy plovers in the continental U.S., and suggest that Puerto Rico snowy plovers should be recognized as part of a distinct subspecies. In 1989, there were at most 20 breeding pairs of snowy plovers in Puerto Rico (Lee 1989) and no known large populations in the Caribbean. The current status of snowy plovers in Puerto Rico is unknown because of a lack of more recent monitoring data, but likely it has not improved substantially. This suggests that this population is at high risk of extinction due to demographic and potentially genetic factors. Genetic variation within this population was also low according to microsatellite heterozygosity and allelic richness which are relatively insensitive to sample size variation among sites (Table 1). MtDNA nucleotide diversity was high in Puerto Rico due to the presence of divergent haplotypes at this site (Table 1), but low heterozygosity at 10 independent microsatellite loci indicates substantial inbreeding nonetheless. Island populations tend to have lower levels of genetic variation than mainland populations due to isolation, small population size, and/or founder effects, thereby increasing extinction risks (Frankham 1997, 1998). Genetic distinctness, extremely small population size, and low genetic variation of the Puerto Rico snowy plovers all suggest that this population should be a high conservation priority.
Management of Pacific Coast snowy plovers should take into consideration the high genetic connectivity between Pacific Coast and Great Basin breeding regions. While these regions are connected by sufficient gene flow to homogenize gene pools (which may only require a few individuals per generation), they may continue to function as demographically independent populations, particularly given their unique habitats, low dispersal rates (Page 2004), and population declines (Page et al. 1991). The likely adaptive genetic variation between Pacific Coast and Great Basin snowy plovers in habitat choice and life histories is important to conserve to allow adaptation to future environmental changes. These differences may not be detectable in patterns of neutral variation at mtDNA and microsatellite loci (McKay and Latta 2002).
Our data do not support recognition of southeastern U.S. snowy plovers as a separate genetic unit. However, they are listed as threatened by the state of Florida and endangered by the state of Alabama because of population declines, habitat degradation due to development, increasing recreational use of beach habitat, and vulnerability to catastrophic events, particularly hurricanes (Chase and Gore 1989; Gore 1996; Sprandel et al. 1997). As with Pacific Coast and Great Basin snowy plovers, there may be adaptive differences between southeastern U.S. snowy plovers, which use coastal habitats, and interior snowy plovers that may not be detected with neutral genetic markers.
Conclusions
Our mtDNA and microsatellite data provide strong evidence for continued recognition of three subspecies of snowy plovers (C. a. nivosus, C. a. tenuirostris, and C. a. occidentalis) in the Western Hemisphere, but suggest changing the geographic boundary for C. a. nivosus and C. a. tenuirostris. Additionally, we found no genetic differences between Pacific Coast and Great Basin snowy plovers, but note that there may still be limited dispersal between these populations from a demographic standpoint, as well as adaptive differences. Similarly, because of likely demographic independence and to preserve potential adaptive variation, we suggest that southeastern U.S. snowy plovers warrant conservation concern, despite minimal genetic differentiation from other continental U.S. breeding areas at neutral loci.
Our approach of combining multiple markers and placing genetic data in the context of field data should also be useful for better defining subspecies and developing conservation priorities for Kentish/snowy plovers in Europe, Asia, and Africa which are also in decline due to many of the same factors affecting snowy plovers in North America and the Caribbean.
ESA protection of subspecies highlights the importance of defining geographic boundaries for subspecies of conservation concern. However, it can be difficult due to differences in opinion on what criteria to use and when data are conflicting. In this study, mtDNA and microsatellite data were concordant, suggesting that there are three subspecies of snowy plovers within the Western Hemisphere. Regardless, using multiple types of data and explicitly defining subspecies criteria prior to analysis will increase confidence in subspecies identification and conservation prioritization.
References
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Automatic Control 19:716–723
Amadon D (1949) The seventy-five percent rule for subspecies. Condor 51:250–258
American Ornithologists’ Union (1957) Check-list of North American birds, 5th edn. American Ornithologists’ Union, Washington, DC
Ball RM, Avise JC (1992) Mitochondrial DNA phylogeographic differentiation among avian populations and the evolutionary significance of subspecies. Auk 109:626–636
Ballard JWO, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13:729–744
Barrowclough TG, Vogler AP (2000) Detecting the geographical pattern of speciation from species-level phylogenies. Am Nat 155:419–434
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Nat Acad Sci USA 98:4563–4568
Benedict NG, Oyler-McCance SJ, Taylor SE, Braun CE, Quinn TW (2003) Evaluation of the eastern (Centrocercus u. urophasianus) and western (Centrocercus urophasianus phaios) subspecies of sage-grouse using mitochondrial control-region sequence data. Conserv Gen 4:301–310
Bhagabati NK, Brown JL, Bowen BS (2004) Geographic variation in Mexican Jays (Aphelocoma ultramarina): local differentiation, polyphyly, or hybridization? Mol Ecol 13:2721–2734
Binford LC (1989) A distributional survey of the birds of the Mexican state of Oaxaca. Ornithol Monogr 43:1–418
Blake ER (1977) Manual of neotropical birds, vol 1. Univ. of Chicago Press, Chicago
Caizergues A, Bernard-Laurent A, Brenot J-F, Ellison L, Rasplus JY (2003) Population genetics structure of rock ptarmigan Lagopus mutus in northern and western Europe. Mol Ecol 12:2267–2274
Chan Y, Arcese P (2002) Subspecific differentiation and conservation of song sparrows (Melospiza melodia) in the San Francisco Bay region inferred by microsatellite loci analysis. Auk 119:641–657
Chase CA III, Gore JA (1989) Snowy plover breeding distribution. Florida Game and Fresh Water Fish Commission, Tallahassee
Clement M, Posada D, Crandall K (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1660
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Coyne JA, Orr HA (2004) Speciation. Sinauer, Sunderland, Massachusetts
Cramp S, Simmons KEL (1983) Handbook of the birds of Europe, the Middle East and North Africa, vol 3. Oxford Univ. Press, New York
Darwin C (1868) The variation of animals and plants under domestication. Murray, London
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Draheim HM (2006) Phylogeography and population structure of least terns (Sterna antillarum). Masters thesis, Oregon State University, Corvallis, Oregon, USA
Eggert LS, Mundy NI, WoodRuff DS (2004) Population structure of loggerhead shrikes in the California Channel Islands. Mol Ecol 13:2121–2133
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Endler JA (1977) Geographic variation, speciation, and clines. Princeton Univ. Press, Princeton, New Jersey
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Frankham R (1997) Do island populations have lower genetic variation than mainland populations? Heredity 78:311–327
Frankham R (1998) Inbreeding and extinction: island populations. Conserv Biol 12:665–675
Frankham R, Ballou JD, Briscoe DA (2002) Resolving taxonomic uncertainties and defining management units. In: Frankham R, Ballou JD, Briscoe DA (eds) Introduction to conservation genetics. Cambridge Univ. Press, United Kingdom, pp 365–392
Fry AJ, Zink RM (1998) Geographic analysis of nucleotide diversity and song sparrow (Aves: Emberizidae) population history. Mol Ecol 7:1303–1313
Futuyma DJ (1998) Evolutionary biology, 3rd edn. Sinauer, Sunderland, Massachusetts
Gaunt AS, Oring LW (1997) Guidelines to the use of wild birds in research. The Ornithological Council, Washington, DC
Gay L, Defos Du Rau P, Mondain-Monval J-Y, Crochet P-A (2004) Phylogeography of a game species: the red-crested pochard (Netta rufina) and consequences for its management. Mol Ecol 13:1035–1045
Given AD, Mills A, Baker AJ (2002). Isolation of polymorphic microsatellite loci from the red-billed gull (Larus novaehollandiae scopulinus) and amplification in related species. Mol Ecol Notes 2:416–418
Gore J (1996) Cuban snowy plover. In: Rodgers H, Kale W, Smith KT (eds) Rare and endangered biota of Florida. V. Birds. University Press of Florida, Gainesville, Florida, pp 61–72
Gorman LR (2000) Population differentiation among snowy plovers (Charadrius alexandrinus) in North America. Masters thesis, Oregon State University, Corvallis, Oregon, USA
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html
Haig SM, Forsman ED, Mullins TD (2004) Subspecies relationships and genetic structure in the spotted owl. Conserv Genet 5:683–705
Haig SM, Beever EA, Chambers SM, Draheim HM, Dugger BD, Dunham S, Elliott-Smith E, Fontaine JB, Kesler DC, Knaus BJ, Lopes IF, Loschl P, Mullins TD, Sheffield LM (2006) Taxonomic considerations in listing subspecies under the U.S. Endangered Species Act. Conserv Biol 20:1584–1594
Haldane JBS (1931) A mathematical theory of natural selection. VI. Isolation. Trans Camb Phil Soc 26:220–230
Hayman P, Marchant J, Prater T (1986) Shorebirds: an identification guide to the waders of the world. Houghton Mifflin Co., Boston
Hill LA, Talent LO (1990) Effects of capture, handling, banding, and radio-marking on breeding least terns and snowy plovers. J Field Ornithol 61:310–319
Huelsenbeck JP, Ronquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Idaghdour Y, Broderick D, Korrida A, Chbel F (2004) Mitochondrial control region diversity of the Houbara bustard Chlamydotis undulata complex and genetic structure along the Atlantic seaboard of North Africa. Mol Ecol 13:43–54
Johnson JA, Toepfer JE, Dunn PO (2003) Contrasting patterns of mitochondrial and microsatellite population structure in fragmented populations of greater prairie chickens. Mol Ecol 12:3335–3347
Jones KL, Krapu GL, Brandt DA, Ashley MV (2005) Population genetic structure in migratory sandhill cranes and the role of Pleistocene glaciations. Mol Ecol 14:2645–2657
Joseph L, Lessa EP, Christidis L (1999) Phylogeny and biogeography in the evolution of migration: shorebirds of the Charadrius complex. J Biogeogr 26:329–342
Kalinowski ST (2002) How many alleles per locus should be used to estimate genetic distances? Heredity 88:62–65
Küpper C, Horsburgh GJ, Dawson DA, Ffrench-Constant R, Székely T, Burke T (2007) Characterization of 36 polymorphic microsatellite loci in the Kentish plover (Charadrius alexandrinus) including two sex-linked loci and their amplification in four other Charadrius species. Mol Ecol Notes 7:35–39
Lafferty KD (2001) Disturbance to wintering western snowy plovers. Biol Conserv 101:315–325
Lee GC (1989) Breeding ecology and habitat use patterns of snowy and Wilson’s plovers at the Cabo Rojo salt flats, Puerto Rico. Master’s thesis, Clemson Univ., Clemson SC
Lewontin R, Krakauer J (1975) Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 74:175–195
Lian C, Zhou Z, Hogetsu T (2001) A simple method for developing microsatellite markers using amplified fragments of inter-simple sequence repeat (ISSR). J Plant Res 114:381–385
Maddison DR, Maddison WP (2000) MacClade 4: analysis of phylogeny and character evolution. Ver 40. Sinauer Associates, Sunderland, Massachusetts
Mayr E (1942) Systematics and the origin of species. Columbia Univ. Press, New York
Mayr E (1963) Animal species and evolution. Harvard Univ. Press, Cambridge, Massachusetts
McKay JK, Latta RG (2002) Adaptive population divergence: markers, QTL and traits. Trends Ecol Evol 17:285–291
Miller MP (2005) Alleles In Space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered 96:722–724
Mills LS, Allendorf FW (1996) The one-migrant-per-generation rule in conservation and management. Conserv Biol 10:1509–1518
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol 19:153–170
Oyler-McCance SJ, St. John J, Knopf FL, Quinn TW (2005) Population genetic analysis of mountain plover using mitochondrial DNA sequence data. Condor 107:353–362
Page GW (2004) Evidence supporting the Pacific coastal population of the western snowy plover as a distinct population segment qualifying for threatened status under the Endangered Species Act. Unpubl. report to the U.S. Fish and Wildlife Service. Point Reyes Bird Observatory Conservation Science, Stinson Beach, California
Page GW, Stenzel LE, Shuford WD, Bruce CR (1991) Distribution and abundance of the snowy plover on its western North American breeding grounds. J Field Ornithol 62:245–255
Page GW, JS Warriner, JC Warriner, Paton PWC (1995) Snowy plover (Charadrius alexandrinus). In: Poole A, Gill F (eds) The birds of North America, vol 154. The Academy of Natural Sciences, Philadelphia, and The American Ornithologists’ Union, Washington, DC
Patten MA, Unitt P (2002) Diagnosability versus mean differences of sage sparrow subspecies. Auk 119:26–35
Paton PWC, Edwards TC Jr (1996) Factors affecting interannual movements of snowy plovers. Auk 113:534–543
Phillimore AB, Owens IPF (2006) Are subspecies useful in evolutionary and conservation biology. Proc Roy Soc Lond B 273:1049–1053
Pitra C, D’Aloia M-A, Lieckfeldt D, Combreau O (2004) Genetic variation across the current range of the Asian Houbara bustard (Chlamydotis undulata macqueenii). Conserv Gen 5:205–215
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Primmer CR, Moller AP, Ellegren H (1995) Resolving genetic relationships with microsatellite markers: a parentage testing system for the swallow Hirundo rustica. Mol Ecol 4:493–498
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Randi E, Tabarroni C, Rimoni S, Lucchini V, Sfougaris A (2003) Phylogeography of the rock partridge (Alectoris graeca). Mol Ecol 12:2201–2214
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Heredity 83:248–249
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Ruhlen TD, Abbott S, Stenzel LE, Page GW (2003) Evidence that human disturbance reduces snowy plover chick survival. J Field Ornithol 74:300–304
Sibley CG, Monroe BL Jr (1990) Distribution and taxonomy of birds of the world. Yale Univ. Press, London
Slatkin M (1985) Gene flow in natural populations. Ann Rev Ecol Syst 16:393–430
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Solorzano S, Baker AJ, Oyama K (2004) Conservation priorities for resplendent quetzals based on analysis of mitochondrial DNA control region sequences. Condor 106:449–456
Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP (1999) Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol Phylogenet Evol 12:105–114
Sprandel GL, Gore JA, Cobb DT (1997) Winter shorebird survey. Final performance report. Florida Game and Freshwater Fish Commission, Talahassee, Florida
Stenzel LE, Warriner JC, Warriner JS, Wilson KS, Bidstrup FC, Page GW (1994) Long-distance breeding dispersal of Snowy Plovers in western North America. J Anim Ecol 63:887–902
Stroud DA, Davidson NC, West R, Scott DA, Haanstra L, Thorup O, Ganter B, Delany S (2004) Status of migratory wader populations in Africa and western Eurasia in the 1990s. Int Wader Stud 15:1–259
Sullivan J, Abdo Z, Joyce P, Swofford DL (2005) Comparing successive approximations and simultaneous optimization approaches to maximum likelihood estimation of phylogeny from DNA sequences. Mol Biol Evol 22:1386–1392
Swofford DL (2000) PAUP*: phylogenetic analysis using parsimony (* and other methods). Sinauer, Sunderland, Massachusetts
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins D (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
USFWS (US Fish and Wildlife Service) (1978) Determination that 11 plant taxa are endangered species and two plant taxa are threatened species. Federal Register 43:17910–17916
USFWS (U.S. Fish and Wildlife Service) (1993) Determination of threatened status for the Pacific Coast population of the western snowy plover. Federal Register 58:12864–12874
USFWS (U.S. Fish and Wildlife Service) and NMFS (National Marine Fisheries Service) (1996) Policy regarding the recognition of distinct vertebrate population segments under the Endangered Species Act. Federal Register 61:4721–4725
Valliantoes M, Lougheed SC, Boag PT (2002) Conservation genetic of the loggerhead shrike (Lanius ludovicianus) in central and eastern North America. Conserv Gen 3:1–13
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Warriner JS, Warriner JC, Page GW, Stenzel LE (1986) Mating system and reproductive success of a small population of polygamous Snowy Plovers. Wilson Bull 98:15–37
Wenink PW, Baker AJ, Tilanus GJ (1994). Mitochondrial control-region sequences in two shorebird species, the turnstone and the dunlin, and their utility in population genetic studies. Mol Biol Evol 11:22–31
Wilson-Jacobs R, Meslow EC (1984) Distribution, abundance, and nesting characteristics of snowy plovers on the Oregon coast. Northwest Sci 58:40–48
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–259
Zink RM (1989) The study of geographic variation. Auk 106:157–160
Zink RM (2004) The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proc R Soc Lond B 271:561–564
Zink RM, Barrowclough GF, Atwood JL, Blackwell-Rago RC (2000) Genetics, taxonomy, and conservation of the threatened California gnatcatcher. Conserv Biol 14:1394–1405
Acknowledgements
We thank A. Archuleta, K. Brennan, S. Cardiff, K. Castelein, M. Colwell, B. Cosler, D. Dittmann, R. Estelle, K. Fahy, J. Gerwin, L. Gorman, S. Hackett, C. Hallet, L. Hanauska, D. Lauten, A. Musche, M.P. Nieto, G. Page, A. Powell, P. Sanzenbacher, M. Stern, R. Swift, O. Taft, J. Van Remsen, J. Whittier, and D. Wilson for collecting tissue samples. Logistical support was provided by Cabo Rojo, Lower Rio Grande Valley, Quivira, and Willapa Bay National Wildlife Refuges, Florida Game and Fresh Water Fish Commission, Florida Division of Recreation and Parks, Gulf Island National Seashore, Eglin Air Force Base, Louisiana Museum of Natural History, Texas Parks and Wildlife Division, Inland Sea Shorebird Reserve, Bureau of Land Management Salt Lake District Office, Utah Division of Wildlife Resources, and Summer Lake Wildlife Area. We are grateful to C. Küpper for sharing primer sequences; B. Bowen, E. Elliott-Smith, S. Oyler-McCance, S. Talbot, and two anonymous reviewers for providing comments on earlier drafts of this paper; P. Martin for discussions regarding subspecies issues; and USFWS (regions 1 and 4), USGS Forest and Rangeland Ecosystem Science Center, and the USGS Science Support Program for providing funding for this work. Views expressed in this paper are the authors’ and do not necessarily reflect the view of the USGS.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Rights and permissions
About this article
Cite this article
Funk, W.C., Mullins, T.D. & Haig, S.M. Conservation genetics of snowy plovers (Charadrius alexandrinus) in the Western Hemisphere: population genetic structure and delineation of subspecies. Conserv Genet 8, 1287–1309 (2007). https://doi.org/10.1007/s10592-006-9278-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-006-9278-7