
Abstract
The present study sought to determine the anti-tumor effects of OXA-11, a potent, novel small-molecule amino pyrimidine inhibitor (1.2 pM biochemical IC50) of focal adhesion kinase (FAK). In studies of cancer cell lines, OXA-11 inhibited FAK phosphorylation at phospho-tyrosine 397 with a mechanistic IC50 of 1 nM in TOV21G tumor cells, which translated into functional suppression of proliferation in 3-dimensional culture with an EC50 of 9 nM. Studies of OXA-11 activity in TOV21G tumor-cell xenografts in mice revealed a pharmacodynamic EC50 of 1.8 nM, indicative of mechanistic inhibition of pFAK [Y397] in these tumors. OXA-11 inhibited TOV21G tumor growth in a dose-dependent manner and also potentiated effects of cisplatin on tumor cell proliferation and apoptosis in vitro and on tumor growth in mice. Studies of pancreatic neuroendocrine tumors in RIP-Tag2 transgenic mice revealed OXA-11 suppression of pFAK [Y397] and pFAK [Y861] in tumors and liver. OXA-11 given daily from age 14 to 17 weeks reduced tumor vascularity, invasion, and when given together with the anti-VEGFR-2 antibody DC101 reduced the incidence, abundance, and size of liver metastases. Liver micrometastases were found in 100 % of mice treated with vehicle, 84 % of mice treated with OXA-11, and 79 % of mice treated with DC101 (19–24 mice per group). In contrast, liver micrometastases were found in only 52 % of 21 mice treated with OXA-11 plus DC101, and those present were significantly smaller and less numerous. Together, these findings indicate that OXA-11 is a potent and selective inhibitor of FAK phosphorylation in vitro and in vivo. OXA-11 slows tumor growth, potentiates the anti-tumor actions of cisplatin and—when combined with VEGFR-2 blockade—reduces metastasis of pancreatic neuroendocrine tumors in RIP-Tag2 mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
FAK, also known as protein tyrosine kinase2 (PTK2, FAK1), is a cytosolic non-receptor tyrosine kinase located at sites of integrin clustering in focal adhesions [1]. FAK is upregulated in many solid tumors, and is considered a potential therapeutic target because of its importance in integrating extracellular signals and initiating downstream signaling that regulates cell proliferation, migration, and survival both in tumor and stromal cells [1–6]. Extensive in vitro studies have shown that cell motility is increased by FAK signaling and impaired by FAK inhibition [7–10]. Much progress has also been made in understanding the role of FAK in tumor progression [11–17], and at least six FAK inhibitors are being tested in clinical trials for cancer (https://clinicaltrials.gov/).Footnote 1 However, the use of FAK inhibitors to reduce tumor invasion and metastasis is at an early stage of understanding.
Inhibitors of VEGF ligand or VEGF receptor-2 (VEGFR-2) can block angiogenesis, slow tumor growth, and improve progression-free survival in multiple types of cancer [18–20]. Effects of these agents on invasion and metastasis are less well understood, and result in more hypoxic and aggressive tumors in some preclinical models [21–25]. FAK is a well-documented downstream target of VEGF receptor signaling [26–28], and concurrent inhibition of FAK and VEGFR-2 has promising effects [29].
In the present study, we characterized the effects of OXA-11, a novel small-molecule amino pyrimidine inhibitor of FAK catalytic activity [30]. OXA-11 was found to inhibit FAK Tyr397 (pFAK [Y397]) in TOV21G cells with a biochemical half-maximal inhibitory concentration (IC50) of 1.2 pM and a cellular mechanistic IC50 of 1 nM. Specificity measurements of cellular mechanistic IC50 for 192 kinases in TOV21G cells revealed that OXA-11 inhibited FAK with eightfold selectivity over PYK2 (protein tyrosine kinase2 beta, PTK2B, FAK2), 26-fold selectivity over RSK1/2 (ribosomal protein S6 kinase, polypeptides 1/2), and >1000-fold selectivity over SRC oncogene, aurora kinase A (AURKA), aurora kinase B (AURKB), insulin receptor (IR, INSR), and KDR (VEGFR-2, FLK1). Functional studies of OXA-11 on TOV21G cell growth and survival in 3-dimensional (3-D) culture gave half-maximal effective concentration (EC50) values of 9 nM for inhibition of proliferation and 31 nM for promotion of apoptosis.
Experiments using TOV21G xenografts in mice revealed dose-dependent tumor growth inhibition (TGI). OXA-11 at a dose of 120 mg/kg daily produced 77 % inhibition of growth and 61 % suppression of pFAK [Y397]. Studies of OXA-11 given together with cisplatin revealed amplification of the anti-proliferative and pro-apoptotic actions of chemotherapy in vitro and TGI in mice. OXA-11 effects on metastasis were examined by administering the agent alone or in combination with the selective anti-VEGFR-2 antibody DC101 [31] to RIP-Tag2 transgenic mice, which spontaneously develop pancreatic neuroendocrine carcinomas that metastasize at later stages [23–25]. OXA-11 did not directly inhibit VEGFR-2, but inhibition of FAK and VEGFR-2 together can have complementary effects [26, 32, 33]. Accordingly, OXA-11 given together with DC101 significantly reduced the incidence, abundance, and size of liver metastases.
Materials and methods
OXA-11 kinase selectivity and cellular activity
Biochemical IC50 values for FAK inhibition by OXA-11 (OSI Pharmaceuticals Inc./Astellas Pharma Inc. [30]) were determined by measuring inhibitory activity on FAK-catalyzed phosphorylation of a peptide substrate using the in vitro FAK Omnia Kinase Assay (Invitrogen). The assay was optimized for GST-tagged full-length FAK enzyme, using a sulfonamido-oxine (SOX)-containing peptide as a substrate for FAK and chelation-enhanced fluorescence as the readout (Invitrogen). IC50 values were calculated from 11-point threefold serial dilution assays, using final OXA-11 concentrations ranging from 0.17 to 10,000 nM or from 0.12 to 7250 nM. Assays were run in triplicate.
The biochemical inhibitory activity of OXA-11 on phosphorylation of FAK was compared to inhibition of 191 other kinases (Supplemental Table 1) using a Kinase Profiling System with ADP-Glo® Kinase Assay technology (Promega). Kinases found to have the highest levels of biochemical inhibition (FAK, PYK2, RSK1/2), as well as those whose activity can confound the interpretation of in vivo efficacy studies (aurora kinase A, aurora kinase B, insulin receptor, KDR), were also analyzed in Omnia Kinase Assays to determine the IC50 values for OXA-11 inhibition in cell-based kinase assays (Table 1). Cellular mechanistic IC50 values for OXA-11 inhibition of FAK in lysates of cells from five species were similarly compared (Table 2). Values were calculated as the average of two replicates.
Effects of OXA-11 on FAK phosphorylation, proliferation, and apoptosis were examined in multiple cell lines (American Type Culture Collection, Manassas, VA). TOV21G, IGROV1, OVCAR3, and OVCAR5 immortalized ovarian adenocarcinoma cells were used because of FAK overexpression in many ovarian cancers [34–39]. MDA-MB-361 human breast cancer cells were also used in some experiments [40]. Cells were grown in 3-D culture in Matrigel (BD Biosciences Lot #19232). OXA-11 inhibition of pFAK [Y397] in lysates of cells exposed to OXA-11 concentrations ranging from 0.01 to 100 nM for 2 h was assayed by sandwich ELISA (Invitrogen). The anti-proliferative activity of OXA-11 at these concentrations on cells in 3-D culture over 7 days was determined by measuring cell number with the CellTiter-Glo® Assay (Promega). Pro-apoptotic activity was assessed by measuring caspase 3/7 activity with the Caspase-Glo® 3/7 Assay (Promega). OXA-11 amplification of cisplatin (0.003–30 µM) effects was tested on TOV21G cell cultures. Data from three independent experiments were analyzed by non-linear regression (GraphPad Prism).
OXA-11 activity on FAK in mouse tumor models
Mice were housed under barrier conditions in the animal care facility at UCSF or OSI Pharmaceuticals Inc./Astellas. Body weight was recorded during treatment. All experimental procedures were approved by the Institutional Animal Care and Use Committee of UCSF or OSI/Astellas according to ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
TOV21G or MDA-MB-361 cell lines were implanted subcutaneously in athymic nude mice and allowed to grow to a standardized size (n = 8 mice/group). For studies of pharmacokinetics (PK), pharmacodynamics (PD), and FAK phosphorylation, mice were given a single dose of OXA-11 (40–120 mg/kg, solubilized in 20 % Trappsol) or 20 % Trappsol alone by gavage in a volume of 5 µL/g body weight. Two to 24 h later, tumor levels of pFAK [Y397] were determined by ELISA, and plasma levels of OXA-11 were measured. For dose–response studies of tumor growth inhibition, mice were given OXA-11 (20–120 mg/kg) daily by gavage for 3 weeks. Tumor size was measured during treatment. In studies of OXA-11 potentiation of actions of chemotherapy, OXA-11 (40–120 mg/kg) and/or cisplatin (5 mg/kg) were given daily for 3 weeks to mice with TOV21G tumors.
RIP-Tag2 transgenic mice (C57BL/6 background), which spontaneously develop pancreatic neuroendocrine tumors that express the SV40 T-antigen oncogene under the insulin promoter [41], were treated with OXA-11 and/or DC101, a rat anti-mouse VEGFR-2 antibody [31], beginning at 14 weeks of age. Maximum tolerated dose was determined by administering OXA-11 daily at a dose of 20–120 mg/kg for 3 weeks (n = 5–10 mice per dose). Vehicle-treated mice received 20 % Trappsol alone (n = 8 mice).
DC101 was purified from the supernatant of a hybridoma culture (AM063-PURE, low endotoxin, no azide, in phosphate-buffered saline (PBS), Cell Culture Facility, UCSF) and was injected ip in a dose of 1 mg in 200 µL twice a week. The maximum tolerated dose of OXA-11 + DC101 was determined by giving DC101 with OXA-11 at daily doses of 20 (n = 10 mice) or 40 mg/kg for 3 weeks (n = 8 mice). Both doses were well tolerated when given alone.
Effects of administration of OXA-11 and/or DC101 to RIP-Tag2 mice were compared in four groups treated for 3 weeks beginning at age 14 weeks: (i) vehicle daily; (ii) OXA-11, 20 mg/kg daily; (iii) DC101, 1 mg ip twice a week; or (iv) OXA-11 + DC101 administered as in the other groups. This experimental design enabled treatment-effects on metastasis to be studied in RIP-Tag2 mice at 17 weeks of age when liver metastases are common. Tissues were prepared 24 h after the final treatment.
Total FAK and pFAK [Y397] were assayed by ELISA in tumors of 14-week old RIP-Tag2 mice that received no treatment or received one dose of OXA-11 (40 mg/kg) or 20 % Trappsol 4 or 8 h earlier. Mice were perfused through the vasculature with PBS prior to tumor removal and freezing. The pancreas of tumor-free RIP-Tag2-negative littermates was used as a control.
The cellular targets and time-course of FAK inhibition by OXA-11 were determined by immunohistochemistry in RIP-Tag2 mice that received one dose of OXA-11 (20 mg/kg). Mice that received 20 % Trappsol served as controls. The magnitude and time-course of effects on FAK phosphorylation after OXA-11 were assessed from changes in pFAK [Y397] and pFAK [Y861] immunoreactivity in tumors and in liver sinusoidal endothelial cells [42]. Based on results of pharmacodynamic studies of OXA-11 in TOV21G tumors, pFAK [Y397] immunoreactivity was examined at 2 h (n = 4 mice per group). pFAK [Y861] was also examined in tumors because FAK phospho-tyrosine 861 is activated by VEGF and phosphorylated by SRC [32, 33] and is blocked by FAK inhibitors [43]. pFAK [Y861] was examined from 1 to 24 h (6 time points, n = 4 mice per time point) to compare the time course to pFAK [Y397].
Tissue preparation for immunohistochemistry and imaging
After treatment, mice used for immunohistochemistry were anesthetized with ketamine (100 mg/kg, ip) plus xylazine (10 mg/kg, ip) and perfused through the heart with 1 % paraformaldehyde (PFA) in PBS. The pancreas and liver were removed, fixed for 1 h in PFA at 4 °C, washed three times with PBS, incubated overnight in 30 % sucrose, embedded in OCT, and frozen at −80 °C.
Cryostat sections were stained with two or three primary antibodies to label the following structures by immunohistochemistry: (i) FAK (rabbit anti-mouse FAK, 1:200, Millipore #04-591); (ii) phosphorylated FAK (polyclonal rabbit anti-pFAK [Y397], 1:100, Abcam #AB4803; or polyclonal rabbit anti-pFAK [Y861], 1:200, Invitrogen #44626G); (iii) tumor cells (polyclonal rabbit anti-SV40 T-antigen, 1:500, Santa Cruz Biotechnology #SC-20800; or guinea pig anti-swine insulin, 1:100, Dako #A0564); (iv) endothelial cells (hamster anti-mouse CD31, 1:500, Thermo Scientific #MA3105; rat anti-mouse CD31, 1:500, BD Pharmingen #553370; goat anti-mouse VEGFR-2, 1:500, R&D Systems #AF644; goat anti-mouse podocalyxin, 1:500, R&D Systems #AF1556; or rat anti-mouse CD105, 1:200, eBioscience #14-1051-82); (v) basement membrane and extracellular matrix (rabbit anti-mouse type IV collagen, 1:2000, CosmoBio Co. #LSL-LB-1403-EX; or polyclonal chicken anti-laminin, 1:500, Abcam #AB14055); (vi) acinar pancreas (polyclonal rabbit anti-amylase, 1:500, Sigma #A8273-1VL). Sections 80-µm in thickness were used throughout to visualize the vasculature of tumors and liver in 3-dimensions, while permitting thin optical sections to be obtained from the same tissues by confocal microscopy [44].
Primary antibodies were identified by one or more of 13 secondary antibodies that included goat anti-guinea pig, anti-rabbit, anti-Armenian hamster, anti-chicken, or anti-rat IgG and donkey anti-rabbit, anti-rat, anti-chicken, or anti-goat IgG fluorescently labeled by FITC, Cy3, or Cy5 (Jackson ImmunoResearch; all diluted 1:500). Cell nuclei were stained with Vectashield mounting medium containing DAPI (Vector Laboratories). Specimens were examined with a Zeiss Axiophot fluorescence microscope equipped with a low-light, externally cooled, three-chip charge-coupled device camera (CoolCam; SciMeasure Analytical Systems, Atlanta, GA) and with a Zeiss LSM 510 confocal microscope [44].
Analysis of FAK phosphorylation, tumor size, vascularity, and invasiveness
FAK phosphorylation was analyzed in a digital image of one representative 240 by 240 µm region of tissue obtained from each mouse by confocal microscopy with standardized settings (40× objective). Images were converted to 8-bit grayscale (ImageJ), and a histogram was generated to show the proportion of pixels at each fluorescence intensity in the range of 0–255. A threshold fluorescence intensity of 50 was found to distinguish image foreground from background, and the proportion of pixels above this threshold was used to calculate the fractional area of staining (area density) for pFAK [Y397] or pFAK [Y861]. The overall mean ± standard error of the mean (SE) for each treatment group (n = 4 mice per group) was calculated from values from each mouse.
The effect of treatment on tumor size and vascularity was assessed in sections stained for SV40 T-antigen and CD31 (n = 6 mice per group, except n = 5 for OXA-11). Images of all tumors visible in a section were captured (5× objective, 1× Optovar, 1 pixel = 16 µm2). Tumors too large to fit in a single image were recorded as multiple images, and montages were made with Photoshop. Sectional area of each tumor (average of four tumors per mouse) was measured with ImageJ. Tumor vascularity was expressed as the area density of CD31-positive pixels. Invasiveness was assessed in images of sections of the same tumors stained for amylase and insulin [23]. Invasiveness was assessed by measuring the amylase-positive cells that were completely surrounded by insulin-positive tumor cells and was expressed as the total area of intratumoral amylase-positive pixels per tumor. This approach made it possible to assess invasiveness independently of tumor size, as the two variables were influenced differently by the treatments. Group means were calculated from mean values per mouse.
Analysis of liver metastasis
Liver metastases were identified by SV40 T-antigen staining in 80-µm sections cut from liver specimens measuring 5 × 5 × 3 mm. Two of 4–6 sections per slide with the most SV40 T-antigen-positive cells were analyzed for each mouse in four groups: vehicle, n = 23 mice; OXA-11, n = 19 mice; DC101, n = 24 mice; OXA-11 + DC101, n = 21 mice. Microscopic liver metastases (micrometastases) were defined as two or more adjacent SV40 T-antigen-positive cells, with the understanding that some could be cell clusters and not clonal expansions. Intravascular and extravascular tumor cells that met these criteria were included. Micrometastases in sections were counted, the major and minor axes were measured (10× objective, 1.6× Optovar), and the average of the two was used as an index of the size. Single tumor cells were counted separately. The area of each section was measured (ImageJ) in montages of 6–60 images (5× objective, 1× Optovar, 1 pixel = 16 µm2). Values for the two sections from each mouse were combined.
Statistical analysis
Most values are presented as mean ± SE. Data that were not normally distributed are presented in box-and-whisker plots, showing the first quartile (Q1), median, third quartile (Q3), interquartile range (IQR), and distribution tails, or in cumulative frequency curves. In box plots, the upper whisker extends from the third quartile (Q3) to Q3 + (1.5 × IQR) or the maximum value, whichever is smaller; the lower whisker extends from Q1 to Q1 − (1.5 × IQR) or the minimum value, whichever is greater. Differences between groups were analyzed by analysis of variance, except for values for liver metastases where the Kruskal–Wallis test or the Kolmogorov–Smirnov two-sample test was used. A probability value of <0.05 was considered statistically significant.
Results
FAK inhibitory activity in biochemical and cell-based assays
OXA-11 is an amino pyrimidine of novel structure (Fig. 1a) [30]. The chemical name is diethyl (3-methoxy-4-{[4-({2-methyl-7-[trans-4-(4-methylpiperazin-1-yl) cyclohexyl]-3-oxo-2,3-dihydro-1H-isoindol-4-yl}amino)-5-(trifluoromethyl)pyrimidin-2-yl] amino}benzyl) phosphonate. The formula weight is 1048.12. OXA-11 was found to be a potent (1.2 pM biochemical IC50) and highly selective inhibitor of FAK (Supplemental Table 1) that in cellular mechanistic assays inhibited pFAK [Y397] with an IC50 of 1.13 ± 0.28 nM in TOV21G cell lysates (Fig. 1b, c). In 3-D culture, OXA-11 inhibited tumor cell proliferation with an EC50 of 9 nM for TOV21G cells and 0.3 nM for IGROV1 cells (Fig. 1c, d), and promoted tumor cell apoptosis with an EC50 of 31 nM for TOV21G cells and 0.5 nM for IGROV1 cells (Fig. 1e, f).

OXA-11 structure and FAK inhibitory activity. a The chemical structure of OXA-11 [30]. b Summary of IC50 of OXA-11 inhibition of pFAK [Y397] in TOV21G ovarian cancer cell lysates and EC50 of inhibition of proliferation and induction of apoptosis of TOV21G cells in 3-D culture (data in c, e). Also shown are the pharmacodynamic (PD) EC50 and EC90 of OXA-11 inhibition of pFAK [Y397] in TOV21G tumors in mice (data in Fig. 2c, d). c Dose–response of OXA-11 inhibition of pFAK [Y397] and proliferation of TOV21G cells in vitro, where the IC50 of pFAK inhibition was 1.13 ± 0.28 nM. d Corresponding values for OXA-11 inhibition of pFAK [Y397] and proliferation of IGROV1 ovarian cancer cells in vitro, where the IC50 of pFAK inhibition was 0.8 nM. e, f Dose–response of OXA-11 inhibition of proliferation and promotion of apoptosis (caspase 3/7 activity) of TOV21G cells and IGROV1 cells in vitro. Data from 3 independent experiments
Specificity screening of OXA-11 inhibitory activity in biochemical assays of kinase phosphorylation revealed that FAK and 8 of 191 other kinases were inhibited more than 50 % at an OXA-11 concentration of 100 nM (Supplemental Table 1). However, corresponding cellular mechanistic studies revealed that the IC50 value for OXA-11 was 10 nM for FAK and 80 nM for PYK2, but was 260 nM for RSK1/2 and was >10 µM for insulin receptor, SRC, KDR (VEGFR-2), and the other kinases tested (Table 1).
Comparison of cells from five species revealed that OXA-11 inhibited FAK with an IC50 of 8 nM in mouse 4T1 cells, 20 nM in human MDA-MB-361 cells, and at slightly higher values in rat, dog, and monkey cells (Table 2). Comparison of eight human tumor cell lines revealed that high baseline pFAK [Y397] level was generally predictive of response to OXA-11. TOV21G, OVCAR3, OVCAR5, and IGROV1 cells had relatively high baseline pFAK [Y397] and EC50 values of 0.3–14.5 nM for inhibition of proliferation in 3-D culture, but four other cell lines with lower baseline pFAK [Y397] values had EC50 values >100 nM (Fig. 2a).

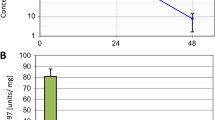
Dose-response, pharmacokinetics (PK), and pharmacodynamics (PD) of OXA-11 inhibition of pFAK [Y397]. a Higher baseline tumor cell pFAK [Y397] was predictive of greater response to OXA-11 in eight ovarian cancer cell lines. Four tumor cell lines with the highest pFAK [Y397] levels had the lowest EC50 for OXA-11 inhibition of proliferation in vitro. b OXA-11 inhibition of TOV21G tumor growth over the dosage range of 20–120 mg/kg daily for 3 weeks. c, d PK/PD analysis of OXA-11 inhibition of pFAK [Y397] in TOV21G tumors in mice after one 80-mg/kg dose (c). Comparison of plasma exposure and inhibition of pFAK [Y397] at 4, 8, and 24 h after one OXA-11 dose of 80 or 120 mg/kg (d). e OXA-11 PD in MDA-MB-361 breast tumors in mice reflected by inhibition of pFAK [Y397] over 24 h after one dose of 40 or 120 mg/kg (n = 8 mice/group). OXA-11 inhibited MDA-MB-361 tumor growth over 3 weeks by 60 % at the lower daily dose and 95 % at the higher dose (data not shown)
FAK inhibitory activity in mouse tumor models
Dose–response studies of OXA-11 inhibition of tumor growth revealed efficacy over a dosage range of 20–120 mg/kg daily for 3 weeks, with 22–67 % reduction in size of TOV21G tumors in nude mice (Fig. 2b). No dose-limiting toxicity was found in athymic nude mice in this dosage range and duration. Pharmacodynamic EC50 measurements of pFAK [Y397] inhibition in TOV21G tumors at 4, 8, and 24 h gave values of 1.8 nM for free and 102 nM for total OXA-11 (Fig. 2c). The half-life of pFAK [Y397] inhibition in TOV21G tumors was about 4 h after a single dose of OXA-11 (80 or 120 mg/kg) (Fig. 2d). pFAK [Y397] was reduced by approximately 25 % at 24 h, when plasma levels had returned to baseline (Fig. 2d). Studies of OXA-11 inhibition of pFAK [Y397] in MDA-MB-361 breast tumors in mice with gave similar results (Fig. 2e).
RIP-Tag2 mice were more sensitive to OXA-11. Daily OXA-11 doses of 40 or 60 mg/kg were well tolerated for at least 3 weeks (Fig. 3a), but higher doses were not. pFAK [Y397] activity in RIP-Tag2 tumors was more than twice the value in normal pancreas and was significantly reduced at 4 h after a single 40-mg/kg dose of OXA-11 (Fig. 3b, c). RIP-Tag2 mice that received DC101 with OXA-11 at 40 mg/kg had weight loss and impaired survival beyond 2 weeks of treatment, but DC101 given with OXA-11 at a daily dose of 20 mg/kg was well tolerated (Fig. 3d).

OXA-11 tolerability and inhibition of pFAK Tyr397 and Tyr861 in RIP-Tag2 mice. a OXA-11 safety at doses of 40 or 60 mg/kg daily for 3 weeks, beginning at age 14 weeks (n = 8-10 mice per group). b ELISA assay of FAK and pFAK [Y397] in untreated tumors over a range of total protein loaded in samples of constant volume (n = 5 mice per group), which revealed pFAK [Y397] levels in the range found in TOV21G tumors. BQL, below quantifiable limit. c Comparison of pFAK [Y397] activity in RIP-Tag2 tumors at baseline (vehicle), 4 or 8 h after one dose of OXA-11 (40 mg/kg), and in the normal pancreas of wild-type mice (n = 4–12 mice per group). d Survival of RIP-Tag2 mice during treatment to compare OXA-11 doses of 20 and 40 mg/kg given with DC101 for 3 weeks, beginning at age 14 weeks (n = 9–10 mice per group). When given with DC101, the lower dose was well tolerated, but the higher dose was not. e–g Comparison of effect of one dose of vehicle (e) or OXA-11 (f) on suppression of pFAK [Y397] staining (red) in liver of 14-week old RIP-Tag2 mice treated 2 h before fixation, and corresponding measurements (g). The value at 2 h was 86 % less than baseline. h–j Comparison of effect of vehicle or OXA-11 on suppression of pFAK [Y861] (red) in liver of mice treated 2 h before fixation (h, i), and measurements from 1 to 24 h after treatment (j). pFAK [Y861] immunoreactivity was significantly less at 1, 2, and 4 h after OXA-11. pFAK immunoreactivity data from images of liver of n = 4 mice/group. *P < 0.05 compared to vehicle, ANOVA. Scale bar in i applies to all images: 40 µm
Studies of FAK phosphorylation in liver of RIP-Tag2 mice revealed that pFAK [Y397] and pFAK [Y861] were restricted to endothelial cells of hepatic sinusoids. pFAK [Y397] immunofluorescence in liver sinusoids was significantly less at 2 h after one 20-mg/kg dose of OXA-11 (Fig. 3e, f). The value at 2 h was 86 % less than baseline (Fig. 3g). Total FAK immunoreactivity did not change (data not shown). pFAK [Y861] in liver sinusoids was similarly reduced (Fig. 3h, i). pFAK [Y861] staining was reduced 79 % at 1 h, 57 % at 4 h, and 35 % at 6 h after a single dose of OXA-11 (20 mg/kg), and gradually returned to baseline over 24 h (Fig. 3j). The reduction in pFAK [Y861] fit with a tissue half-life of OXA-11 inhibition of about 4 h, which was similar to the value for pFAK [Y397] in TOV21G tumors (Fig. 2d).
Immunofluorescence for pFAK [Y397] and pFAK [Y861] was strong in RIP-Tag2 tumors under baseline conditions, where both were located in tumor cells and tumor blood vessels. As in liver sinusoids, staining for pFAK [Y397] (Fig. 4a, b) and pFAK [Y861] (Fig. 4c, d) was reduced to nearly undetectable levels at 2 h after one 20-mg/kg dose of OXA-11. Tumors in RIP-Tag2 mice treated for 3 weeks and then prepared 24 h after the final 20-mg/kg dose had strong FAK [pY397] immunoreactivity (data not shown).

OXA-11 suppression of pFAK in RIP-Tag2 tumors. pFAK [Y397] and pFAK [Y861] immunoreactivities (red) in tumor cells (insulin, green) and blood vessels (CD31, white) in tumors of 14-week old RIP-Tag2 mice 2 h after one 20-mg/kg dose of vehicle or OXA-11. a, b Comparison of pFAK [Y397] after vehicle (a) or OXA-11 (b). c, d Comparison of pFAK [Y861] after vehicle (c) or OXA-11 (d). pFAK [Y397] and pFAK [Y861] (red) are strong in both tumor cells (green) and endothelial cells (white) at baseline but are faint after OXA-11. Representative images from tumors of n = 4 mice/group. Scale bar in a applies to all images: 40 µm
Changes in tumor vascularity, size, and invasiveness
Treatment with OXA-11 and/or DC101 reduced the vascularity of RIP-Tag2 tumors (Fig. 5a). The reduction was greatest when the two agents were given together. Sleeves of vascular basement membrane persisted after tumor blood vessels regressed (Fig. 5b), as reported previously for inhibitors of VEGFR-2 [44]. Sleeves of type IV collagen were less abundant after treatment with OXA-11 + DC101 (Fig. 5b). Tumor invasion into the acinar pancreas, indicated by islands of amylase-positive pancreatic acinar cells surrounded by tumor cells, was also reduced by treatment (Fig. 5c).

Treatment-related effects on tumor blood vessels and invasion. Fluorescence microscopic images of tumors in RIP-Tag2 mice treated for 3 weeks beginning at age 14 weeks. a Tumor blood vessels (CD31, red) are more abundant at baseline (vehicle) than after OXA-11, DC101, or OXA-11 + DC101. b Similarly, sleeves of vascular basement membrane (type IV collagen, green) are more numerous in tumors after vehicle than after OXA-11, DC101, or the combination. c Pancreatic acinar cells (amylase, green) surrounded by tumor cells (insulin, red) are more abundant after vehicle than after the other treatments. d Treatment-related differences in tumor vascularity expressed as fractional area of CD31 staining (n = 5–6 mice/group). Tumor vascularity after OXA-11 + DC101 was significantly less than after vehicle or DC101 alone. e Treatment-related differences in tumor invasion expressed as percent area of intratumoral acinar cells and tumor size expressed as sectional area relative to the value for the vehicle group (n = 5–6 mice/group). P < 0.05 compared to *vehicle or †DC101, ANOVA. Scale bar in a applies to all images: 200 µm
Measurements revealed 58 % reduction in tumor blood vessels after OXA-11 + DC101, compared to reductions of 45 or 36 % after OXA-11 or DC101 alone, respectively (Fig. 5d). The reduction in tumor invasion was greater after OXA-11 (81 %) or OXA-11 + DC101 (79 %) than after DC101 (61 %) (Fig. 5e). Tumors were significantly smaller after DC101 alone or together with OXA-11. Reduction in size was greater after DC101 (71 %) than after OXA-11 (53 %) or the combination (57 %) (Fig. 5e). As a reflection of greater reduction in tumor size after DC101 but greater reduction in invasion after OXA-11, the ratio of invasion to size was less after OXA-11 or OXA-11 + DC101 than after DC101 alone (Fig. 5e).
Reduction in metastasis
Liver metastases were visible at autopsy in a small proportion of 17-week old RIP-Tag2 mice after treatment for 3 weeks. They were most numerous in the vehicle group (18 % of 23 mice), and were about half as frequent after treatment with OXA-11 (10 % of 19 mice), DC101 (8 % of 24 mice), or OXA-11 + DC101 (9 % of 21 mice). Despite the infrequency of grossly visible metastases, micrometastases were found in 100 % of mice in the vehicle group. The 1429 liver micrometastases analyzed in this group were located inside hepatic blood vessels or in the liver parenchyma (Fig. 6a). Some micrometastases in the liver parenchyma were vascularized (Fig. 6b). The largest were located near central veins. Intravascular clusters of tumor cells were most commonly located inside central veins (Fig. 6c). Some micrometastases lacked interior blood vessels but were surrounded by endothelial cells (Fig. 6d). It was unclear whether these were intravascular tumor cell clusters that grew in situ into metastases.

OXA-11 and/or DC101 effect on liver metastases. a–d Confocal microscopic images show the diversity of micrometastases in liver of vehicle-treated RIP-Tag2 mice at age 17 weeks. Tumor cells (SV40 T-antigen, red) and endothelial cells (VEGFR-2, green; CD105, white) are stained. a Low magnification view of liver with multiple micrometastases (red) surrounded by dense network of sinusoids (green), which are prominent in this 80-µm thick section. b Micrometastasis with numerous small blood vessels in liver parenchyma. c Intravascular cluster of tumor cells within a central vein, which unlike portal veins, had strong CD105 staining. d Micrometastasis lacking interior blood vessels but surrounded by endothelial cells. e Differences in incidence of micrometastases in four treatment groups. f Box-and-whisker plots of quartiles (median at red/green junction) showing treatment differences in number of micrometastases/10 mm2 of liver section. For both e and f, values for OXA-11 + DC101 group were significantly less than for vehicle group (*P < 0.01, Kruskal–Wallis test). g Large treatment-related differences in proportion of mice where micrometastases were absent (0 % in vehicle group and 48 % of mice in OXA-11 + DC101 group) or were unusually abundant (>15/100 mm2: 26 % of vehicle group and 5 % of OXA-11 + DC101 group) (*P < 0.05, Kolmogorov–Smirnov two-sample test). h, i Cumulative frequency distributions showing smaller size of micrometastases in OXA-11 + DC101 group (red) than in vehicle group (blue) (h) or DC101 group (green) (i). In contrast, micrometastases in OXA-11 group (magenta) were similar in size to those in vehicle group (j). Black curves show difference between the other two curves. *P < 0.01, Kolmogorov–Smirnov two-sample test. k, l Single tumor cells (red) within liver sinusoids, identified by strong staining for laminin (green) and podocalyxin (white), in vehicle-treated mice. m Box-and-whisker plots showing treatment differences in single tumor cells/10 mm2 of liver. P < 0.05 compared to *vehicle or †OXA-11, Kruskal–Wallis test. Data in e from n = 19–23 mice/group; data in f–j and m from n = 16–21 mice/group. Scale bar in a applies to all images: 200 µm in a, 40 µm in b–d, and 25 µm in k, l
Compared to 100 % incidence of micrometastases in the vehicle group, the incidence was only 52 % in mice treated with OXA-11 + DC101 (Fig. 6e). The incidence was about 80 % in mice treated with either OXA-11 or DC101 alone (Fig. 6e). The median number of micrometastases, expressed per area of liver section, was 85 % less in the OXA-11 + DC101 group than in the vehicle group (Fig. 6f). Similarly, only 5 % of mice in the OXA-11 + DC101 group had abundant micrometastases (>15/100 mm2 liver), in contrast to 26 % of mice in the vehicle group (Fig. 6g). Micrometastases in mice treated with OXA-11 + DC101 were smaller than with vehicle (Fig. 6h) or DC101 (Fig. 6i). The size in mice treated with OXA-11 alone was about the same as in the vehicle group (Fig. 6j).
Single tumor cells were found in liver sinusoids (Fig. 6k, l) of 81 of 87 mice in all treatment groups but varied in abundance with the treatment (Fig. 6m). Single tumor cells were least numerous in the OXA-11 + DC101 group where the number, expressed per area of liver section, was 88 % less than in the vehicle group (Fig. 6m).
Micrometastases in the vehicle group were more heterogeneous (Figs. 6a–d, 7a) than those in the other groups. Intravascular micrometastases were especially common in the OXA-11 group (Fig. 7b). By comparison, more micrometastases in the DC101 group had dense vascularization, irregular borders, and scattered tumor cells (Fig. 7c, arrows). Micrometastases in the OXA-11 + DC101 group were smaller and more circumscribed, were poorly vascularized and, like those in the OXA-11 group, were commonly located in or near a central vein (Fig. 7d, asterisk).

Treatment-related differences in liver micrometastases. Confocal microscopic images of micrometastases stained for tumor cells (SV40 T-antigen, red) alone (left) or with endothelial cells (VEGFR-2, white) and basement membrane matrix (laminin, green) (right) in liver of 17-week old RIP-Tag2 mice treated for 3 weeks. a Micrometastasis with densely packed tumor cells and punctate laminin matrix but no blood vessels. Tumor-cell cluster is surrounded by vessels and matrix (vehicle-treated mouse). b Micrometastasis located within branches of a hepatic central vein (OXA-11-treated mouse). c Highly vascularized micrometastasis composed of loosely clustered and scattered tumor cells (arrows) (DC101-treated mouse). d Extravascular cluster of densely packed tumor cells with few blood vessels (white) and little matrix (green) located next to a central vein (*) (OXA-11 + DC101-treated mouse). Representative images from tumors of n = 4 mice/group. Scale bar in d applies to all images: 25 µm
OXA-11 potentiation of cisplatin activity
In light of evidence that genetic deletion of FAK in endothelial cells increases the sensitivity of tumor cells to doxorubicin chemotherapy [45], we asked whether pharmacologic inhibition of FAK by OXA-11 can similarly increase the efficacy of the DNA-crosslinking chemotherapeutic cisplatin. Because OXA-11 suppressed endothelial-cell pFAK, we first compared the activity of OXA-11 and/or cisplatin on tumor cell proliferation in culture, to assess the direct tumor-cell effects. OXA-11 plus cisplatin inhibited TOV21G cell proliferation in vitro significantly more than either agent alone (Fig. 8a). Dose–response studies revealed reductions over a broad range of cisplatin concentrations (Fig. 8b). OXA-11 also amplified cisplatin-induced apoptosis of TOV21G cells in vitro (Fig. 8c), and the amplification increased with OXA-11 concentration (Fig. 8d). Studies in mice with implanted TOV21G tumors confirmed that the combination of agents was well tolerated (OXA-11, 40–120 mg/kg, cisplatin, 5 mg/kg). These experiments showed that cisplatin given alone increased pFAK [Y397] in TOV21G-cell tumors, but OXA-11 offset this increase and resulted in a dose-dependent reduction in pFAK [Y397] (Fig. 8e). Cisplatin administered with OXA-11 at 120 mg/kg inhibited growth of TOV21G tumors by 92 % over 3 weeks (Fig. 8f).

OXA-11 potentiation of effects of cisplatin on TOV21G tumor cells in culture and on tumors implanted subcutaneously in nude mice. a Greater inhibition of TOV21G cell proliferation in vitro by cisplatin together with OXA-11 than by either agent alone. b OXA-11 potentiation of anti-proliferative action of cisplatin on TOV21G cells in culture over a broad range of concentrations. c OXA-11 amplification of cisplatin-induced apoptosis of TOV21G cells. d Amount of amplification of apoptosis increased with OXA-11 concentration. e Cisplatin given alone increased pFAK [Y397] in TOV21G-cell tumors in mice, but co-administration of OXA-11 with cisplatin offset this increase and resulted in a dose-dependent reduction in pFAK [Y397]. P < 0.01 compared to *vehicle or †cisplatin alone, two-way ANOVA, n = 8 mice per group. f Cisplatin (5 mg/kg) and OXA-11 (120 mg/kg) both inhibited growth of TOV21G tumors when given as single agents for 3 weeks but resulted in greater inhibition (92 %) when given together (n = 8 mice per group)
Discussion
The present studies had four objectives: (i) determine the IC50/EC50 of OXA-11 inhibition of pFAK [Y397] in tumor cells in vitro and in tumors in mice; (ii) test the efficacy of OXA-11 in reducing tumor cell proliferation and increasing apoptosis in 3-D culture and in slowing tumor growth in mice; (iii) determine the effects of OXA-11 alone or in combination with VEGFR-2 blockade on invasion and metastasis of pancreatic neuroendocrine tumors in RIP-Tag2 mice; and (iv) learn whether OXA-11 potentiates the anti-tumor effects of cisplatin in vitro and in vivo. The experiments revealed the selective inhibitory action of OXA-11 on pFAK [Y397] in tumor cell lysates with 1 nM IC50. The most responsive of 8 tumor cell lines had high baseline levels of FAK phosphorylation. OXA-11 suppressed tumor cell proliferation with 9 nM EC50, promoted apoptosis with 31 nM EC50, and resulted in dose-dependent slowing of tumor growth. OXA-11 administered alone reduced tumor vascularity and, when combined with VEGFR-2 blockade, reduced tumor size, invasion, and incidence, number, and size of liver metastases in RIP-Tag2 mice. OXA-11 also potentiated effects of cisplatin on TOV21G tumor cell proliferation and apoptosis and on tumor growth. The findings document that OXA-11 potently inhibits FAK phosphorylation in tumors, reduces tumor growth, decreases liver metastasis from pancreatic neuroendocrine tumors when coupled with a VEGFR-2 inhibitor, and increases the anti-tumor action of cisplatin.
OXA-11 kinase selectivity
Biochemical assays of 192 kinases indicated that FAK and eight other kinases were inhibited >50 % by OXA-11 at 100 nM. However, cell-based assays of the same kinases revealed that only FAK (IC50 10 nM) and PYK2 (IC50 80 nM) were inhibited by OXA-11 with IC50 values of <100 nM. Although PYK2 can compensate for the absence of FAK [42, 46, 47], OXA-11 should inhibit such effects. Despite OXA-11 potency and selectivity for FAK and PYK2, as with other small-molecule kinase inhibitors, contributions of off-target effects cannot be excluded.
OXA-11 reduction in FAK phosphorylation
The time course of OXA-11 inhibition of pFAK [Y397] in vivo was assessed in TOV21G and MDA-MB-361 tumors in nude mice, where the pharmacodynamic half-life was dose-dependent, in the range of 4–6 h, and phosphorylation remained somewhat below baseline for 24 h. OXA-11 inhibition of pFAK [Y397] was also evaluated in RIP-Tag2 tumors, where FAK was located both in tumor cells and in endothelial cells, and in liver sinusoidal endothelial cells that naturally have high FAK expression. Both pFAK [Y397] and pFAK [Y861] were significantly reduced 2 h after one 20-mg/kg dose of OXA-11. Although pFAK [Y397] in RIP-Tag2 tumors was near baseline at 24 h after the final dose of the 3-week treatment period, the action of OXA-11 on some tumors could be prolonged by accumulation over time.
Proliferation of some ovarian cancer cell lines was not inhibited by OXA-11. These cells had lower baseline pFAK [Y397] levels than cells responsive to OXA-11. Additional factors including stromal contributions are also likely to influence the link between pFAK [Y397] inhibition and slowing of tumor growth and metastasis. The FAK/PYK2 inhibitor PF-562,271 is reported to reduce pFAK [Y397] without slowing growth of several ovarian tumor cells, including SKOV3, OVCAR10, and IGROV1-IP cells in vitro and 5009-MOVCAR cells in mice [37]. Despite this feature, PF-562,271 decreases metastatic spread of these tumors [37], as it does in an ovarian cancer model (ID8-IP) where tumor size and pFAK [Y397] are reduced [17].
OXA-11 had no dose-limiting toxicity over the range of 20–120 mg/kg daily in xenograft-bearing athymic nude mice, but tolerable dosing was limited to 20–60 mg/kg in RIP-Tag2 mice. When given with DC101, OXA-11 had a maximum tolerated dose of 20 mg/kg. Nonetheless, the efficacy of OXA-11 under these conditions was documented by greater reduction in tumor vascularity and incidence, number, and size of liver metastases than found with DC101 alone. The metabolic effects of insulin-secreting pancreatic neuroendocrine tumors are known to influence drug tolerability in RIP-Tag2 mice [48].
Suppression of tumor growth
FAK inhibitors slow tumor growth in some mouse models [8, 9, 15, 16, 49–51]. OXA-11 had this effect on TOV21G tumors. Although tumors in RIP-Tag2 mice treated with OXA-11 tended to be smaller, the reduction was not significant unless DC101 was added. Even then, the reduction in size was about the same as found with DC101 given alone [25, 52], even though the anti-metastatic action of the combination was significantly greater. Of relevance, FAK inhibitors reduce tumor cell dissemination without reducing growth of some tumors [53], perhaps because growth involves the non-kinase domain of FAK, independent of the kinase activity [50, 54].
OXA-11 caused greater reduction in tumor vascularity when given together with DC101. This augmented action is consistent with stronger antivascular effects of the FAK/PYK2 inhibitor PF-562,271 when given with sunitinib to block VEGFR-2 and related kinases [29]. Both VEGFR-2 and FAK play important roles in endothelial cell function and tumor angiogenesis [26, 32, 33, 42], and inhibition of either kinase has well-documented anti-angiogenic actions in tumor models [8, 25, 55]. Although pFAK [Y397] is the standard readout of FAK catalytic activity, pFAK [Y861] is also involved in downstream effects of VEGF signaling [32, 33].
Some of the complementary actions of OXA-11 given together with DC101 could reflect dual effects of blocking FAK directly and as a downstream target of VEGFR-2 signaling [26, 32, 33]. Activation of VEGF signaling through VEGFR-2 promotes endothelial cell migration through a phosphoinositide 3-kinase (PI3-kinase)-dependent mechanism that requires FAK activation [26]. Endothelial-specific FAK knockout mice have impaired VEGF-driven angiogenesis and developmental vascular defects [28, 42, 56–58]. Although not directly inhibited by DC101, VEGFR-3 is expressed on some tumor blood vessels and contributes to tumor angiogenesis [59] and could be implicated through direct interaction with FAK [60].
To build on the finding of complementary effects of blocking FAK and VEGF-driven angiogenesis together, we sought to determine whether OXA-11 increased the efficacy of chemotherapy. Previous work has shown that genetic deletion of FAK in endothelial cells increases the sensitivity of tumor cells to doxorubicin chemotherapy [45]. We found that OXA-11 amplified the tumor growth-slowing action of cisplatin. OXA-11 also prevented the increase in FAK found in TOV21G tumors treated with cisplatin alone, and caused a dose-dependent decrease in pFAK [Y397]. Consistent with a direct action on tumor cells, OXA-11 potentiated the anti-proliferative and pro-apoptotic effects of cisplatin in vitro. Therefore, unlike the essential involvement of endothelial cell-FAK in increased tumor-cell sensitivity to doxorubicin chemotherapy [45], OXA-11 action on blood vessels was not required for amplification of cisplatin effects on tumor cells.
Suppression of liver metastasis
FAK inhibitors have been found to reduce metastasis in multiple tumor models [15–17, 37]. The multistage nature of tumor progression in RIP-Tag2 mice, where growth and spread occur through natural routes and mechanisms, made the model particularly useful for studies of FAK inhibition in liver metastasis. The liver is the principal organ for hematogenous metastasis in these mice, because venous blood from the pancreas drains through the hepatic portal system. Also, liver metastases—even single tumor cells—were readily identified by staining for SV40 T-antigen. Although 100 % of 23 mice in the vehicle group had liver micrometastases, only 52 % of 21 mice treated with OXA-11 together with DC101 had micrometastases, and those present were smaller and less numerous.
The response of metastases to angiogenesis inhibitors can be influenced by co-option of existing blood vessels, which reduces the dependence of tumor growth on angiogenesis [61–63]. In the present study, micrometastases that were located within or near blood vessels could reflect vessel co-option. Micrometastases in most mice were circumscribed, had little or no blood supply, and were located in or near central veins. DC101-treated mice were an exception, where about 30 % of the larger micrometastases were well-vascularized and invaded the surrounding liver tissue. DC101 is known to have pro-metastatic effects in RIP-Tag2 mice [21, 25]. Although DC101 did not increase the incidence or abundance of liver metastases in the present study, it was accompanied by better vascularized and more invasive metastases.
The abundance of intravascular tumor cells in the liver indicated a high incidence of intravasation of these cells in RIP-Tag2 mice. OXA-11-driven reduction in FAK phosphorylation in tumor cells and liver sinusoids could change the adhesive interactions necessary for extravasation and metastasis [64]. Inhibition of FAK can also change endothelial barrier function, prevent VEGF-induced leakage, and tumor cell transmigration [65, 66]. Complementary actions of OXA-11 and VEGFR-2 blockade on tumor cells and endothelial cells could contribute to the favorable effects on invasion and metastasis.
Conclusions
The findings demonstrate that OXA-11 is a potent and selective inhibitor of FAK phosphorylation in tumor cells in vitro and in vivo. When combined with VEGFR-2 blockade, OXA-11 reduces liver metastases from pancreatic neuroendocrine tumors. OXA-11 also potentiates the anti-tumor actions of cisplatin. The results highlight the potential benefit of combining FAK inhibitors with antiangiogenic agents or chemotherapy.
Notes
VS-6063/PF-04554878 (Verastem), GSK2256098 (GlaxoSmithKline), PF-00562271 (Pfizer), VS-4718 (Verastem), BI-853520 (Boehringer Ingelheim), CEP-37440 (Teva Pharmaceuticals).
References
Golubovskaya VM (2014) Targeting FAK in human cancer: from finding to first clinical trials. Front Biosci 19:687–706
Parsons JT (2003) Focal adhesion kinase: the first ten years. J Cell Sci 116:1409–1416
Gabarra-Niecko V, Schaller MD, Dunty JM (2003) FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev 22:359–374
Sood AK, Coffin JE, Schneider GB, Fletcher MS, DeYoung BR, Gruman LM, Gershenson DM, Schaller MD, Hendrix MJ (2004) Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am J Pathol 165:1087–1095
McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC (2005) The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer 5:505–515
Sulzmaier FJ, Jean C, Schlaepfer DD (2014) FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer 14:598–610
Mitra SK, Hanson DA, Schlaepfer DD (2005) Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6:56–68
Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, Lorenzen M, Berliner M, Luzzio M, Patel N, Schmitt E, LaGreca S, Jani J, Wessel M, Marr E, Griffor M, Vajdos F (2008) Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res 68:1935–1944
Bagi CM, Roberts GW, Andresen CJ (2008) Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastases. Cancer 112:2313–2321
Schaller MD (2010) Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 123:1007–1013
Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M, Schlaepfer DD, Alaoui-Jamali MA (2005) FAK signaling is critical for ErbB-2/ErbB-3 receptor cooperation for oncogenic transformation and invasion. J Cell Biol 171:505–516
Li S, Dong W, Zong Y, Yin W, Jin G, Hu Q, Huang X, Jiang W, Hua ZC (2007) Polyethylenimine-complexed plasmid particles targeting focal adhesion kinase function as melanoma tumor therapeutics. Mol Ther 15:515–523
Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, Muller WJ (2007) Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci USA 104:20302–20307
Wendt MK, Schiemann WP (2009) Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res 11(1–16):R68
Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, Stupack DG, Schlaepfer DD (2010) Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther 9:778–790
Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, Stelow EB, Parsons JT, Bauer TW (2011) Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther 10:2135–2145
Ward KK, Tancioni I, Lawson C, Miller NL, Jean C, Chen XL, Uryu S, Kim J, Tarin D, Stupack DG, Plaxe SC, Schlaepfer DD (2013) Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp Metastasis 30:579–594
Ferrara N, Mass RD, Campa C, Kim R (2007) Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med 58:491–504
Aprile G, Rijavec E, Fontanella C, Rihawi K, Grossi F (2014) Ramucirumab: preclinical research and clinical development. Onco Targets Ther 7:1997–2006
Ciombor KK, Berlin J (2014) Aflibercept—a decoy VEGF receptor. Curr Oncol Rep 16:368
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15:220–231
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15:232–239
Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman HA, Christensen JG, Aftab DT, McDonald DM (2012) Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov 2:270–287
Sennino B, Ishiguro-Oonuma T, Schriver BJ, Christensen JG, McDonald DM (2013) Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res 73:3692–3703
Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, Bussolino F, Serini G, Giraudo E (2012) Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest 122:1832–1848
Qi JH, Claesson-Welsh L (2001) VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp Cell Res 263:173–182
Masson-Gadais B, Houle F, Laferriere J, Huot J (2003) Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones 8:37–52
Lechertier T, Hodivala-Dilke K (2011) Focal adhesion kinase and tumour angiogenesis. J Pathol 226:404–412
Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, Tuthill T, Andresen CJ (2009) Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther 8:856–865
Appari RD, Chen X, Chilukuri R, Crew AP, Dong H, Ferraro C, Foreman K, Gupta RC, Li A-H, Sherman D, Stolz KM, Volk B, Zahler R (2013) Amino pyrimidine anticancer compounds. Patent No: US008399433B2. Issued by: United States Patent and Trademark Office. Issue Date: March 19, 2013. Assignee: OSI Pharmaceuticals, LLC, Farmingdale, pp 1–310
Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Bohlen P (1998) Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev 17:155–161
Abu-Ghazaleh R, Kabir J, Jia H, Lobo M, Zachary I (2001) Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem J 360:255–264
Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA (2002) Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol 157:149–160
Provencher DM, Lounis H, Champoux L, Tetrault M, Manderson EN, Wang JC, Eydoux P, Savoie R, Tonin PN, Mes-Masson AM (2000) Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell Dev Biol Anim 36:357–361
Karam AK, Santiskulvong C, Fekete M, Zabih S, Eng C, Dorigo O (2010) Cisplatin and PI3kinase inhibition decrease invasion and migration of human ovarian carcinoma cells and regulate matrix-metalloproteinase expression. Cytoskeleton (Hoboken) 67:535–544
Tonin PN, Hudson TJ, Rodier F, Bossolasco M, Lee PD, Novak J, Manderson EN, Provencher D, Mes-Masson AM (2001) Microarray analysis of gene expression mirrors the biology of an ovarian cancer model. Oncogene 20:6617–6626
Shah NR, Tancioni I, Ward KK, Lawson C, Chen XL, Jean C, Sulzmaier FJ, Uryu S, Miller NL, Connolly DC, Schlaepfer DD (2014) Analyses of merlin/NF2 connection to FAK inhibitor responsiveness in serous ovarian cancer. Gynecol Oncol 134:104–111
Yoon H, Choi YL, Song JY, Do I, Kang SY, Ko YH, Song S, Kim BG (2014) Targeted inhibition of FAK, PYK2 and BCL-XL synergistically enhances apoptosis in ovarian clear cell carcinoma cell lines. PLoS ONE 9:e88587
He Y, Wu AC, Harrington BS, Davies CM, Wallace SJ, Adams MN, Palmer JS, Roche DK, Hollier BG, Westbrook TF, Hamidi H, Konecny GE, Winterhoff B, Chetty NP, Crandon AJ, Oliveira NB, Shannon CM, Tinker AV, Gilks CB, Coward JI, Lumley JW, Perrin LC, Armes JE, Hooper JD (2015) Elevated CDCP1 predicts poor patient outcome and mediates ovarian clear cell carcinoma by promoting tumor spheroid formation, cell migration and chemoresistance. Oncogene. doi:10.1038/onc.2015.101
Sutherland RL, Hall RE, Pang GY, Musgrove EA, Clarke CL (1988) Effect of medroxyprogesterone acetate on proliferation and cell cycle kinetics of human mammary carcinoma cells. Cancer Res 48:5084–5091
Hanahan D (1985) Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315:115–122
Tavora B, Batista S, Reynolds LE, Jadeja S, Robinson S, Kostourou V, Hart I, Fruttiger M, Parsons M, Hodivala-Dilke KM (2010) Endothelial FAK is required for tumour angiogenesis. EMBO Mol Med 2:516–528
Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G, Sood AK (2007) Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res 67:10976–10983
Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM (2004) Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 165:35–52
Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, Lees DM, Wong PP, Alexopoulou A, Elia G, Clear A, Ledoux A, Hunter J, Perkins N, Gribben JG, Hodivala-Dilke KM (2014) Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 514:112–116
Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA, Chen XL, Gothert JR, Shen TL, Guan JL, Schlaepfer DD, Cheresh DA (2008) Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol 181:43–50
Kostourou V, Lechertier T, Reynolds LE, Lees DM, Baker M, Jones DT, Tavora B, Ramjaun AR, Birdsey GM, Robinson SD, Parsons M, Randi AM, Hart IR, Hodivala-Dilke K (2013) FAK-heterozygous mice display enhanced tumour angiogenesis. Nat Commun 4(1–11):2020
Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM (2011) Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res 71:1573–1583
Kurio N, Shimo T, Fukazawa T, Okui T, Hassan NM, Honami T, Horikiri Y, Hatakeyama S, Takaoka M, Naomoto Y, Sasaki A (2012) Anti-tumor effect of a novel FAK inhibitor TAE226 against human oral squamous cell carcinoma. Oral Oncol 48:1159–1170
Golubovskaya VM, Figel S, Ho BT, Johnson CP, Yemma M, Huang G, Zheng M, Nyberg C, Magis A, Ostrov DA, Gelman IH, Cance WG (2012) A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3,5,7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 33:1004–1013
Golubovskaya VM, Huang G, Ho B, Yemma M, Morrison CD, Lee J, Eliceiri BP, Cance WG (2013) Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol Cancer Ther 12:162–172
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8:299–309
Brunton VG, Frame MC (2008) Src and focal adhesion kinase as therapeutic targets in cancer. Curr Opin Pharmacol 8:427–432
Lim ST, Mikolon D, Stupack DG, Schlaepfer DD (2008) FERM control of FAK function: implications for cancer therapy. Cell Cycle 7:2306–2314
Stone RL, Baggerly KA, Armaiz-Pena GN, Kang Y, Sanguino AM, Thanapprapasr D, Dalton HJ, Bottsford-Miller J, Zand B, Akbani R, Diao L, Nick AM, DeGeest K, Lopez-Berestein G, Coleman RL, Lutgendorf S, Sood AK (2014) Focal adhesion kinase: an alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer Biol Ther 15:919–929
Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL (2005) Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol 169:941–952
Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R (2006) Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 172:151–162
Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH (2003) Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res 92:300–307
Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K (2008) Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454:656–660
Gogate PN, Ethirajan M, Kurenova EV, Magis AT, Pandey RK, Cance WG (2014) Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein–protein interaction. Eur J Med Chem 80:154–166
Stessels F, Van den Eynden G, Van der Auwera I, Salgado R, Van den Heuvel E, Harris AL, Jackson DG, Colpaert CG, van Marck EA, Dirix LY, Vermeulen PB (2004) Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J Cancer 90:1429–1436
Zhao C, Yang H, Shi H, Wang X, Chen X, Yuan Y, Lin S, Wei Y (2011) Distinct contributions of angiogenesis and vascular co-option during the initiation of primary microtumors and micrometastases. Carcinogenesis 32:1143–1150
Donnem T, Hu J, Ferguson M, Adighibe O, Snell C, Harris AL, Gatter KC, Pezzella F (2013) Vessel co-option in primary human tumors and metastases: an obstacle to effective anti-angiogenic treatment? Cancer Med 2:427–436
von Sengbusch A, Gassmann P, Fisch KM, Enns A, Nicolson GL, Haier J (2005) Focal adhesion kinase regulates metastatic adhesion of carcinoma cells within liver sinusoids. Am J Pathol 166:585–596
Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD (2012) VEGF-induced vascular permeability is mediated by FAK. Dev Cell 22:146–157
Jean C, Chen XL, Nam JO, Tancioni I, Uryu S, Lawson C, Ward KK, Walsh CT, Miller NL, Ghassemian M, Turowski P, Dejana E, Weis S, Cheresh DA, Schlaepfer DD (2014) Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol 204:247–263
Acknowledgments
We gratefully acknowledge the important contributions by Elisabeth Buck, Andrew Crew, and Maryland Franklin in performing the experiments with OXA-11 at OSI Pharmaceuticals. We also thank OSI for supplying OXA-11 for the studies performed at UCSF. The authors thank Maximilian Nitschké and Barbara Sennino for critical review of the manuscript and Brian Schriver for genotyping the RIP-Tag2 mice at UCSF. This work was supported in part by National Heart, Lung, and Blood Institute grants HL24136, HL59157, and HL127402 from the US National Institutes of Health (to DMcD), funding from OSI and AngelWorks Foundation (to DMcD), and an Overseas Research Fellowship from University of Bergen, Norway (to IM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest to disclose. DE is a former employee of OSI Pharmaceuticals.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Moen, I., Gebre, M., Alonso-Camino, V. et al. Anti-metastatic action of FAK inhibitor OXA-11 in combination with VEGFR-2 signaling blockade in pancreatic neuroendocrine tumors. Clin Exp Metastasis 32, 799–817 (2015). https://doi.org/10.1007/s10585-015-9752-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9752-z