
Abstract
Galectin-1 is a β-galactoside-binding protein overexpressed by cancer cells. The primary roles of galectin-1 in cancer progression and metastasis are attributed to suppression of T cell immune responses, promotion of tumor angiogenesis and increased tumor cell adhesion and invasion. Using pulmonary metastasis models of murine breast (4T1) and colon (CT26) cancer, we demonstrate that targeting galectin-1 with thiodigalactoside (TDG) or shRNA galectin-1 knockdown (G1KD) results in a significant reduction in lung metastasis. Increased numbers of CD4+ helper T cells and CD8+ cytotoxic T lymphocytes were found in the peripheral blood of both TDG-treated and G1KD cell challenged mice. The levels of TUNEL+ apoptotic cancer cells and the presence of CD3+ T cells were also increased in lung metastases. Furthermore, galectin-1 was found to bind to the adhesion molecules, CD44 and CD326, which are also known as markers of breast and colon cancer stem cells, and TDG likely blocks galectin-1 binding to these molecules. The TDG-mediated inhibition of galectin-1 binding reduced 4T1 cell adhesion to the basement membrane protein laminin, Matrigel and EAhy926 endothelial cell surfaces. These findings establish possible mechanisms for the anti-metastatic effect of galectin-1 inhibition and suggest that targeting galectin-1 may represent a promising and effective anti-metastatic therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The occurrence of metastasis is the primary cause of morbidity and mortality of cancer patients. Lung is a frequent metastatic target for a variety of cancers, including breast, colorectal, melanoma, renal, pancreatic and sarcoma [1, 2]. In order to develop an immunotherapeutic approach for inhibiting metastatic diseases, it is important to promote the levels of CD8+ cytotoxic T lymphocytes (CTLs) capable of effectively killing tumor cells through antigen-specific responses. A number of pre-clinical and clinical studies have evaluated immunotherapies against metastasis and demonstrated that increased levels of CTLs in primary tumors and circulation is required early on to prevent metastasis [3, 4]. However, the avidity of T cells is often not potent enough to completely control metastatic diseases [5]. This is probably due to the numerous ways which tumor cells use to evade the immune surveillance systems [6–10]. Therefore, overcoming these immunosuppressive mechanisms will also be essential to improve outcomes of anti-metastatic immunotherapies.
In addition to immune modulation, the other focus of metastasis treatment is to inhibit cancer cell adhesion. The interaction of circulating cancer cells with the pulmonary endothelium is a critical first step required for metastatic cells to colonize the lung. This is facilitated by two possible mechanisms [11]. The first mechanism involves physically arresting and retaining cancer cells in the lung capillaries. Circulating cancer cells are often aggregated with cells and platelets, resulting in clogging of the lung capillaries, followed by infiltration and colonization. Secondly, adherence of circulating cancer cells to the pulmonary endothelial cells is promoted by various surface adhesion molecules. For example, integrins (e.g., α4, α6β1, αvβ3), tumor-associated glycoproteins (e.g., MUC1), epithelial cell adhesion molecule (EpCAM, CD326) and CD44 expressed on cancer cells can bind to a number of endothelial surface molecules, such as intercellular adhesion molecule-1 (ICAM-1, CD54), platelet–endothelial cell adhesion molecule-1 (PECAM-1, CD31), vascular cell adhesion molecule-1 (VCAM-1, CD106) or selectins [11, 12]. In addition, CD44 variant isoforms (CD44v) and CD326 have been demonstrated to support metastatic extravasation by promoting cell adhesion [13, 14]. Interestingly, CD44 and CD326 have also been recognized as surface markers of pancreatic, breast and colon cancer stem cells (CSCs) that may possess a high metastatic capacity [15–17].
Galectins are soluble β-galactoside binding lectin families overexpressed by a range of cancer cells [18, 19]. The galectin carbohydrate recognition domains (CRDs) are highly conserved and capable of recognizing galactosides (e.g., Galβ1-3GlcNAc and Galβ1-4GlcNAc) [20]. Among the 15 mammalian galectins, galectin-1, -3 and -9 have been established to play important roles in cancer biology and progression [19]. In particular, galectin-1 is well known for its roles in suppressing effector T cell functions by inducing apoptosis in activated T cells [21–24] and promoting regulatory T cell functions [25]. Galectin-1 is also involved in multiple processes of cancer pathology, such as tumor cell aggregation [26], cell migration [27, 28], cell adhesion [29, 30] and promotion of angiogenesis [31–33]. Banh et al. [22] have recently reported that metastasis of Lewis lung carcinoma was significantly reduced by shRNA galectin-1 knockdown (G1KD). However, detailed mechanisms for targeting galectin-1 effects on reducing metastasis are yet to be clearly demonstrated. In the present study, the mechanisms whereby targeting galectin-1 suppresses lung metastasis were explored using a galectin CRD inhibitor, thiodigalactoside (TDG) [34], as well as G1KD in both 4T1 breast cancer and CT26 colon cancer murine models. The results showed a significant reduction in lung metastases after treatment with TDG or when G1KD cells were utilized to induce metastases. CD4+ helper T (Th) cells and CD8+ CTLs in the peripheral blood were significantly increased in the TDG-treated and G1KD cell challenged groups. The levels of TUNEL+ apoptotic cancer cells in the lung metastases were enhanced with increased levels of CD3+ T cells. In addition, it was found that galectin-1 binds to adhesion molecules, CD44 and CD326, which are important to initiate metastasis. TDG-mediated inhibition of galectin-1 binding significantly reduced cancer cell adhesion to endothelial substratums. These findings provide evidence that galectin-1 inhibition suppresses lung metastasis by protecting effector T cells and inhibiting adhesion of cancer cells. Thus, galectin-1 may represent a promising and effective target for anti-metastatic therapy.
Materials and methods
Cell lines and mouse experiments
Murine breast cancer 4T1, 4T1.2 mCherry+ (obtained from the Peter MacCallum Cancer Institute, Melbourne) and murine colon cancer CT26 were cultured in DMEM supplemented with 10 % heat-inactivated fetal bovine serum (FBS), 50 IU/ml penicillin and streptomycin, 20 mM HEPES, and 1.6 mM l-glutamate. Galectin-1 expression in tumor cells was knocked down by lentiviral transfection of pLKO.1-puro-LGALS1 [32]. Non-targeting shRNA control (shRNA NC) vector was used in each experiment. EAhy926 endothelial cells were grown in DMEM containing 10 % FBS and HAT supplement (Invitrogen). Female Balb/c mice at 5–6 weeks of age were obtained from the Animal Resources Centre (Western Australia). 4T1, 4T1.2 or CT26 (5 × 105) cells were injected into the tail vein of Balb/c mice. Saline or TDG (10 mg/kg) was administrated i.v. simultaneously and continued every second day. On day 15, the mice were sacrificed and lung metastases were evaluated by lung weight, immunohistochemistry, flow cytometry and by counting the number of surface metastatic foci.
Immunohistochemistry of lung tissues
Excised lung tissues were frozen in O.C.T. compound before sectioning into 15–20 μm using cryostat and transferred onto SuperFrost slides (MENZEL-GLÄSER). The lung sections were fixed and permeabilized by ice-cold acetone and incubated for 15 min. After washing with 0.025 % Triton X 100/PBS (TPBS), the tissue sections were blocked with 3 % bovine serum albumin (BSA)/TPBS for 1 h in a humid chamber, followed by incubation with primary antibodies for 1 h. After washing with TPBS, the sections were incubated with fluorophore-conjugated secondary antibodies for 1 h in the dark. After washing with TPBS, nuclei were counter-stained with VECTASHIELD mounting reagent containing 4′-6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Primary antibodies used were; biotinylated anti-mouse galectin-1 goat polyclonal IgG (R&D Systems), anti-mouse galectin-1 rat monoclonal IgG (R&D systems, clone: 201002), anti-mouse CD44 rat monoclonal IgG (Abcam, clone: IM7), anti-mouse CD326 (EpCAM) rabbit monoclonal IgG (Abcam, clone: E144), anti-mouse CD3 rat IgG (eBioscience, clone: 17A2) and Alexa Fluor 488 anti-mouse CD31 rat IgG (BioLegend, clone: MEC13.3). Secondary antibodies used were, Alexa Fluor 488 anti-rat IgG, Alexa Fluor 488 anti-rabbit IgG, Alexa Fluor 594 anti-rat IgG (donkey host, Invitrogen) and PE streptavidin (Jackson ImmunoResearch). The slides were analyzed with the Ti-Eclipse epifluorescence (Ti-E) microscope and NIS-Elements software (Nikon).
Flow cytometric analysis and galectin-1 binding assay
For 4T1.2 mCherry+ metastasis analysis, excised lung tissues were dissociated by treatment with 70 U/ml of collagenase (Sigma) at 37 °C for 1 h. The percentage of mCherry+ cells were analyzed by the LSRFortessa flow cytometer with FACSDiva software (BD Biosciences). Blood was collected by cardiac puncture and peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Paque Plus (GE Healthcare). Fc receptor (FcR) was blocked by incubation with anti-mouse CD16/CD32 IgG (Innovex Biosciences) at 4 °C for 5 min, followed by immunostaining with fluorophore-conjugated antibodies at 4 °C for 20 min in the dark. The antibodies used were; eFluor 450 anti-mouse CD3ε rat IgG, FITC anti-mouse CD4 rat IgG (eBioscience) and PE anti-mouse CD8a rat IgG (BD Pharmingen). The cells were fixed in 1 % p-formaldehyde/PBS and analyzed By the LSRFortessa. For galectin-1 competition studies, cancer cells were pre-incubated with anti-CD44 rat IgG (IM7), anti-CD326 rabbit IgG (E144) or matched isotype control IgG at 4 °C for 30 min. After washing with 1 % FBS/PBS, recombinant galectin-1 (1 μg/ml) was added with or without TDG (0.1 mg/ml). After washing, the cells were incubated with biotinylated anti-mouse galectin-1 goat IgG, followed by incubation with PE streptavidin or Alexa Fluor 647 anti-goat IgG (donkey host, Invitrogen). The level of surface bound galectin-1 was analyzed using the LSRFortessa. Data were analyzed using FlowJo software to overlay histograms.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) Click-iT assay
TUNEL assay was performed using the Click-iT TUNEL Alexa Fluor 488 imaging kit (Invitrogen) following the manufacturer’s protocol. Briefly, the tissue sections (8–10 μm) were fixed and permeabilized by ice-cold acetone for 15 min. The TdT reaction cocktail was added and incubated for 1 h, followed by 30 min incubation with the Click-iT reaction cocktail. The tissues were then blocked in 3 % BSA/PBS for 1 h at room temperature. After washing, the tissue sections were covered by VECTASHIELD mounting reagent containing DAPI. The slides were analyzed with the Ti-E microscope.
Purification of recombinant galectin-1
Recombinant human galectin-1 was purified as previously described [32]. In brief, BL21 DE3 Escherichia coli transformed with pET-3a-hgal-1 was amplified and lysed. Galectin-1 protein was purified by affinity chromatography using a lactose-Sepharose column. Purified recombinant galectin-1 was dialyzed at 4 °C for 4 h and the buffer exchanged three times with PBS containing 4 mM 2-mercaptoethanol and 5 mM ethylenediaminetetraacetic acid (EDTA).
Cell sorting
A 4T1 cell suspension was incubated with anti-CD44 rat IgG or anti-CD326 rabbit IgG at 4 °C for 30 min, followed by Alexa Fluor 488 anti-rat IgG or AlexaFluor 594 anti-rabbit IgG in the dark. After washing, the stained 4T1 cells were sorted based on the levels of surface CD44 or CD326 using the MoFlo XDP and Summit Software (Beckman Coulter). The sorted cells were cultured for 2–3 days and used for galectin-1 binding assay.
Immunoprecipitation and western blot analysis
4T1 cell lysates were prepared with ice-cold modified radioimmunoprecipitation (RIPA) buffer [150 mM NaCl, 50 mM Tris–HCl (pH 7.4), 0.25 % sodium deoxycholate, 1 mM NaF, 10 μg/ml Leupeptin, 1 mM PMSF]. After centrifuging at 14,000 rpm at 4 °C for 5 min, the supernatants were subjected to a pre-clearing step with protein A/G Agarose beads (Thermo Fisher Scientific) at 4 °C for 30 min. Supernatants obtained by a brief centrifugation at 2,000 rpm were treated with recombinant galectin-1 (1 μg/ml) and incubated at 4 °C for 1 h, followed by incubation with anti-galectin-1 goat IgG (R&D Systems) or IgG isotype control at 4 °C for 3 h. The immunoprecipitates were then incubated with protein A/G Agarose beads at 4 °C overnight. The beads were washed three times with modified RIPA buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (2×) [0.125 M Tris–Cl (pH 6.8), 2 % SDS, 25 μg/ml bromophenoll blue R250, 5 % β-ME and 20 % glycerol] was then added, followed by incubation at 98 °C for 3 min. The samples were resolved in 12.5 % SDS-PAGE gels, transferred to polyvinylidene fluoride (PVDF) membrane, and immunoblotted with anti-CD44 rat IgG or anti-CD326 rabbit IgG. The secondary antibodies used were horseradish peroxidase (HRP)-conjugated goat anti-rat IgG (Bio-rad) or goat anti-rabbit IgG (Cell Signaling). The 4T1 whole cell lysate was used as control. Blots were developed with Supersignal West Pico Substrate (Thermo Fisher Scientific) and detected by ChemiDoc XRS system (Bio-rad).
Immunofluorescence
4T1 cells were cultured in a 48-well plate and fixed by 1 % p-formaldehyde/PBS, followed by washing with PBS. After incubation with FcR blocker at 4 °C for 5 min, the cells were pre-incubated with anti-CD44 rat IgG or isotype control at 4 °C for 30 min. After washing, recombinant galectin-1 (1 μg/ml) was added with or without TDG (0.1 mg/ml) and incubated in 1 % BSA/PBS at 4 °C for 30 min. After washing, the cells were incubated with biotinylated anti-galectin-1 IgG at 4 °C for 30 min, followed by incubation with PE streptavidin at 4 °C for 30 min. The nuclei were counter-stained by SYTOX Green (Invitrogen). The cells were fixed by 1 % p-formaldehyde/PBS and analyzed by the Ti-E microscope.
Cell adhesion assay
4T1 cells were labeled using Vybrant DiI (Invitrogen, Em; 565 nm) and pre-incubated with anti-CD44 rat IgG, anti-CD326 rabbit IgG or matched isotype IgG at 4 °C for 30 min. After washing, galectin-1 (1 μg/ml) was added with or without TDG (0.1 mg/ml) and incubated at 37 °C for 30 min. Natural mouse laminin (Invitrogen) was used to coat 24-well plates by incubation at 4 °C overnight in serum free DMEM. Monolayer of EAhy926 endothelial cells was prepared on 24-well plates. BD Matrigel Basement Membrane Matrix (BD Biosciences) (50 μl) was added to μ-Slides Angiogenesis (ibidi), followed by incubation at 37 °C for 30 min. The DiI labeled cancer cell suspension (1 × 106 cells/ml in 1 % FBS/PBS) was added onto either laminin, Matrigel or EAhy926 cell coated plates. After the plate was incubated at 37 °C for 10–30 min, non-adherent cells were removed by gentle washing with pre-warmed 1 % FBS/PBS. Adherent cells were then counted and imaged using the Ti-E microscope. For EAhy926 containing wells, in order to detect the DiI labeled cancer cells, total cells were detached by incubation with 10 mM EDTA/PBS and DiI+ cells were detected by the LSRFortessa.
Statistical analysis
Values are expressed as mean ± SEM, and the number (n) of samples used was as indicated. The statistical significance of differences between experimental and control groups was determined by Student’s t test with p values considered significant; *p < 0.05, **p < 0.01, ***p < 0.001. All statistical analyses were performed using GraphPad Prism v4.03.
Results
Anti-metastatic effects of targeting galectin-1 in murine models
To assess whether targeting galectin-1 would affect murine lung metastasis, the synthetic disaccharide TDG, which binds to galectin CRDs, or shRNA G1KD cell lines were utilized [32]. The galectin-1 expression levels by each cell line, including shRNA NC, are shown in Supplementary Fig. 1a and revealed about 50 % reduction in levels in the galectin-1 specific shRNA treated, pooled cell population. We first confirmed that there was no significant difference in cell growth between WT and G1KD 4T1.2 or CT26 cells (Supplementary Fig. 1b, c), which is consistent with previous reports [22, 32]. Next, WT, G1KD or shRNA NC 4T1.2 mCherry+ or CT26 cells (5 × 105) were injected i.v. into the tail of Balb/c mice, followed by saline or TDG (10 mg/kg) administrated i.v. every second day. In the CT26 model, lung weight was significantly decreased in the TDG-treated or G1KD group, compared to WT or shRNA NC mice, respectively (Fig. 1a) and this was associated with a reduced number of detectable surface metastatic lesions (Fig. 1b). The lung weights in the 4T1.2 model were also significantly reduced in the TDG-treated or G1KD cell challenged mice (Fig. 1c). Fluorescence microscopy clearly showed notably reduced regions of mCherry+ foci present in the TDG-treated or G1KD lung sections (Fig. 1d). To quantify the ratio of mCherry+ cells in the lung, the population of collagenase treated whole lung cells were analyzed by flow cytometry. The representative dot-plots showing reductions of mCherry+ cells in TDG-treated or G1KD group (Fig. 1e). The ratio of 4T1.2 mCherry+ cells in the lung were approximately 55.5 % reduced in TDG-treated or G1KD groups, compared to WT or shRNA NC challenged mice (Fig. 1f).

Targeting galectin-1 suppresses lung metastasis of 4T1.2 and CT26. WT 4T1.2 mCherry+ or CT26 cells (5 × 105) were injected i.v. into Balb/c mice, followed by saline or TDG i.v. treatment every second day. Alternatively, G1KD 4T1.2 mCherry+ or CT26 cells (5 × 105) were injected i.v. and saline injected i.v. every second day. The mice were sacrificed on day 15. For the CT26 model, a lung weights and b numbers of metastasis foci were analyzed. For the 4T1.2 model, c lung weights were measured. d mCherry+ regions (red) in the lung were detected by immunohistochemistry. The nuclei were counter-stained by DAPI (blue) and the slides were analyzed on the Ti-E microscope (magnification: ×10). e mCherry+ cells in the lung were also analyzed by flow cytometry (x axis: forward scatter, y axis: mCherry fluorescence intensity) and f the percentage of mCherry+ cells were quantified. Data are presented as mean ± SEM if two independent experiments (total n = 5, 6). Significant difference from the WT control is indicated as: *p < 0.05 or **p < 0.01 (by Student’s t test). (Color figure online)
Increased CD4+ and CD8+ T cell responses against lung metastases
One of the major roles of galectin-1 in tumor progression is in suppressing immunosurveillance by inducing apoptosis in the effector T cells [21, 23]. Therefore, it was evaluated whether TDG treatment or G1KD affected the number of CD3+CD4+ Th cells or CD3+CD8+ CTLs in the PBMC populations. The levels of CD3, CD4 and CD8 in the total PBMCs at day 15 were analyzed by flow cytometry. Figure 2a shows that TDG treatment or the use of G1KD cells increased the populations of CD3+CD4+ (yellow) and CD3+CD8+ (blue) T cells in peripheral blood in both 4T1.2 and CT26 lung metastasis models, compared to mice challenged with WT or shRNA NC cancer cells, respectively. Figure 2b, c depicts the levels of CD3+CD4+ or CD3+CD8+ T cells in the peripheral blood between indicated groups in the 4T1 model. When compared to the levels of T cells in normal Balb/c mice, WT or shRNA NC 4T1 cell challenged mice significantly reduced numbers of CD3+CD4+ or CD3+CD8+ T cells. However, the reduced levels of these T cells were recovered by TDG treatment. The G1KD also increased the levels of CD3+CD4+ or CD3+CD8+ T cells. In TDG-treated G1KD 4T1 challenged mice, the increased levels of CD3+CD4+ or CD3+CD8+ T cells were observed, compared to G1KD 4T1 challenged saline treated mice. This may be because TDG blocked residual galectin-1 produced by G1KD cells.

Targeting galectin-1 increases the levels of CD4+ and CD8+ T cells in the peripheral blood. a PBMCs prepared from the 4T1 or CT26 i.v. challenged mice were stained with anti-CD3, -CD4 and -CD8 IgG and then analyzed by flow cytometry. 3-D images were created using the Flow-Jo software (x axis: CD4, y axis: CD3, z axis: CD8). The levels of b CD3+CD4+ and c CD3+CD8+ T cells in the naïve Balb/c, TDG treated, WT 4T1.2 challenged, WT 4T1.2 challenged and TDG treated, G1KD 4T1.2 challenged or G1KD 4T1.2 challenged and TDG treated groups were compared. Student’s t tests were performed (total n = 4–20): *,δ p < 0.05, **p < 0.01, or ***,### p < 0.001 (*vs WT or shRNA NC, #naïve mouse control vs WT, δG1KD vs G1KD + TDG)
In addition, we investigated the impact of targeting galectin-1 on T cell levels in the lung. The results from immunohistochemistry of frozen lung tissue sections showed increased numbers of CD3+ T cells (green) nearby to 4T1.2 mCherry+ regions (red) in both TDG-treated and G1KD cell challenged mice (Fig. 3a). The quantified data by flow cytometry shows a trend of an increase in CD3+ T cells in the lungs from the TDG-treated or G1KD groups, compared to WT or shRNA NC 4T1.2 mCherry+ cell injected group, respectively (Fig. 3b). In addition, the lung sections from the TDG-treated or G1KD tumor challenged mice contained higher levels of TUNEL+ apoptotic cells (green), compared to those from the WT challenged mice (Fig. 3c). It was confirmed that TUNEL+ cells were indeed metastatic mCherry+ 4T1.2 cells (red) using a higher magnification ×60. This data suggests that increased effector T cells may contribute to the suppression of metastasis by inducing apoptosis in cancer cells.

Targeting galectin-1 increases the levels of CD3+ T cells and TUNEL+ cells in lung metastases. a CD3+ cells in the lungs from 4T1.2 mCherry+ cell challenged mice were analyzed by immunohistochemistry. The sections were incubated with anti-CD3 rat IgG, followed by Alexa Fluor 488 anti-rat IgG (green). b Collagenase treated whole lung cells were stained with anti-CD3 rat IgG and Alexa Fluor 488 anti-rat IgG. The samples were analyzed by flow cytometry (total n = 3–6). c Click-iT TUNEL assay was performed using the lungs from 4T1.2 mCherry+ cell challenged mice. The Click-iT-EdUTP incorporated into dsDNA strand breaks were detected by Alexa Fluor 488 azide (green). The nuclei were counter-stained with DAPI (blue) and analyzed by the Ti-E microscope (magnification: ×20 or ×60). (Color figure online)
CD44 and CD326 expression and galectin-1 in lung metastatic lesions
As another possible mechanism for the anti-metastatic effects of targeting galectin-1, we hypothesized that galectin-1 would associate with cell adhesion molecules, such as CD44 and CD326. Firstly, in order to confirm that galectin-1 was indeed expressed in lung metastatic lesions, immunohistochemistry was performed using the lung tissue sections. High levels of galectin-1 expression (red) were found in WT 4T1 and CT26 lung foci, but were absent from the highly vascularized (normal tissue) CD31+ regions (Supplementary Fig. 2). Given that G1KD tumor cells failed to form high levels of lung metastases, galectin-1 expression may be critical for initiation of cancer cell metastasis. It was also found that galectin-1+ (red) WT 4T1 or CT26 lung foci express high levels of CD44 and CD326 (green) (Fig. 4a, b). Furthermore, Fig. 4c shows that CD44+CD326+ cells were highly bound with galectin-1 (red). This implies that galectin-1 may interact with CD44 or CD326 on 4T1 cells.

CD44+ or CD326+ cells in lung metastases are bound with galectin-1. a The expression of CD44 (green) and galectin-1 (red) or b CD326 (green) and galectin-1 (red) in the lung sections from WT 4T1 or CT26 cell challenged mice were analyzed by immunohistochemistry. The nuclei were counter-stained with DAPI (blue) and analyzed by the Ti-E microscope (magnification: ×10). c Surface galectin-1, CD44 and CD326 on the collagenase treated whole lung cells from 4T1 or CT26 challenged mice were analyzed by flow cytometry. Galectin-1+ cells indicated by red (P1) were shown in representative dot plots (x axis: CD44, y axis: CD326). (Color figure online)
CD44 and CD326 represent galectin-1 binding targets
Immunoprepicitation was performed to investigate in vitro if galectin-1 binds to CD44 and CD326 molecules expressed by 4T1 cells. Recombinant galectin-1 (1 μg/ml) treated 4T1 cell lysates were immunoprecipitated with anti-galectin-1 IgG. Western blot analysis of the immunoprecipitates detected CD44 and CD326, indicating that galectin-1 binds to both CD44 and CD326 (Fig. 5a). To determine whether the levels of CD44 or CD326 alters the levels of galectin-1 binding to the 4T1 cell surface, cells were sorted for CD44low or high and CD326low or high expression (Supplementary Fig. 3a) and used in a galectin-1 binding assay. Compared to WT 4T1 cells, the level of bound galectin-1 was reduced in CD44low and CD326low cells, indicating that these molecules are galectin-1 binding targets. However, there was unexpectedly no increase observed in CD44high or CD326high cells. The sorted cells were also incubated with anti-CD44 or anti-CD326 IgG to block these surface markers [35], followed by the addition of recombinant galectin-1. When these blockade antibodies were used, galectin-1 binding to CD44high or CD326high was reduced, whereas no significant changes were observed in CD44low or CD326low cells (Fig. 5b). In each cell group, rat IgG or rabbit IgG was used as an isotype control of anti-CD44 or anti-CD326 IgG, respectively. A reduction in galectin-1 binding was similarly observed in 4T1 WT cells (Fig. 5c). In addition to this observation, treatment with TDG (0.1 mg/ml) alone resulted in an almost complete inhibition of galectin-1 binding. The levels of surface galectin-1 on cultured 4T1 cells were analyzed by immunofluorescence. Reduced galectin-1 levels were observed in the anti-CD44 IgG and TDG treated groups (Fig. 5d). The levels of surface galectin-1 were compared between WT and G1KD cells to determine the influence of endogenous galectin-1 on the levels of surface galectin-1. The addition of recombinant galectin-1 (1 μg/ml) to G1KD 4T1 cells resulted in 25 % reduction in the surface galectin-1 levels, compared to that on the WT cells (Supplementary Fig. 3b). These results suggest that CD44 and CD326 are perhaps binding targets of exogenous galectin-1 and importantly that TDG completely inhibits the capacity of galectin-1 binding to 4T1 cell surfaces.

Galectin-1 interacts with CD44 or CD326 adhesion molecules and TDG inhibits the galectin-1 binding to 4T1 cells. a 4T1 cell lysates were immunoprecipitated with anti-galectin-1 goat IgG or isotype IgG and then immunoblotted using anti-CD44 or anti-CD326 IgG. 4T1 whole cell lysate was used as a control. b CD44high/low or CD326high/low 4T1 cells were pre-incubated with anti-CD44 rat IgG, anti-CD326 rabbit IgG or matched isotype IgG, followed by adding of recombinant galectin-1 (1 μg/ml). After washing, the percentages of galectin-1 bound 4T1 cells were analyzed by flow cytometry. Data are representative of two independent experiments (mean ± SEM). Student’s t tests were performed: *,# or δ p < 0.05, **,## or dd p < 0.01 (*vs WT, #vs isotype control, δvs indicated group). c The levels of galectin-1 bound WT 4T1 cells are shown by 3-D histograms. d Fixed 4T1 cells were pre-incubated with anti-CD44 rat IgG or isotype IgG, followed by galectin-1 adding (1 μg/ml) with or without TDG (0.1 mg/ml). After washing, the cells were treated with biotinylated anti-galectin-1 IgG and PE streptavidin. The nuclei were counter-stained with SYTOX Green and analyzed by the Ti-E microscope (magnification: ×20)
Reduction of cell adhesion to endothelial substratums by TDG
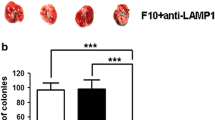
The ability of TDG to block cancer cell adhesion to laminin, Matrigel basement membrane matrix or endothelial cell surfaces as binding substratums by inhibiting galectin-1 binding was evaluated. Fluorescence labeled (DiI, Em; 565 nm) 4T1 cells were added to laminin, Matrigel or EAhy926 cell coated wells and incubated for 10–20 min. After gentle washing, adherent cells were analyzed. Figure 6a shows that cell adhesion to laminin after 20 min incubation was significantly inhibited by pre-incubation with anti-CD44 (p = 0.0012), anti-CD326 (p = 0.0021) IgG or TDG, which displayed the greatest inhibition of cancer cell adhesion to laminin-coated plates (p < 0.001). Figure 6b shows a reduction in Matrigel adherent 4T1 cells following anti-CD44, anti-CD326 IgG or TDG treatment (p = 0.0153–0.0021). Fluorescence microscopy images clearly show visible reduction in DiI+ 4T1 cell binding to the Matrigel following antibody or TDG treatment (Fig. 6c). Flow cytometric analysis (Fig. 6d) demonstrated that blocking CD44 or CD326 with antibody or TDG reduced the binding levels of DiI+ 4T1 cells to EAhy926 endothelial cell surfaces. Figure 6e shows that the amount of EAhy926 adherent 4T1 cells was significantly reduced by anti-CD44 IgG (p = 0.0024) or TDG (p < 0.001). Considered together, the data presented here demonstrate that blocking galectin-1 reduces cell adherence to the endothelial compartments possibly through inhibiting galectin-1 binding to either CD44 or CD326.

TDG inhibits 4T1 cell adhesion to laminin, Matrigel and endothelial cell surfaces. DiI (red) labeled 4T1 cells were incubated with or without anti-CD44 rat IgG or anti-CD362 rabbit IgG or matched isotype IgG. After washing, the cells were incubated with galectin-1 (1 μg/ml) with or without TDG (0.1 mg/ml) for 30 min. The cell samples were added onto either a laminin or b Matrigel coated plates, followed by 20 min incubation at 37 °C. After gentle washing, the numbers of remained labeled 4T1 cells were counted. c Images of the remaining DiI labeled 4T1 cells on laminin or Matrigel were analyzed by the Ti-E microscope (magnification: ×10). d The cell samples were also added onto the EAhy926 cell monolayers in 24-well plates. After co-incubation at 37 °C for 15 min, the total cells were detached and the levels of DiI labeled 4T1 cells were analyzed by flow cytometry. e The percentage of DiI labeled 4T1 cells is shown in the bar graph. f The numbers of cell cluster containing approximately more than 50 cells attached onto Matrigel were counted. Data are representative of two independent experiments (mean ± SEM). Significant difference from the corresponding control is indicated as: *p < 0.05, **p < 0.01 or ***p < 0.001 (by Student t-test)
In addition to the aforementioned cancer cell binding substratums, the formation of aggregated cell clusters following anti-CD44, anti-CD326 IgG or TDG treatment was assessed. In the presence of galectin-1, the number of aggregated cell clusters was considerably increased, whereas anti-CD44, anti-CD326 IgG or TDG treatment reduced the extent of cell aggregation (Fig. 6f). Since cell aggregation increases the chance of tumor cell clogging in the capillaries, this data implies that reduction of cell aggregation by blocking galectin-1 is possibly one mechanism resulting in reduced lung metastasis in TDG-treated mice.
Discussion
Galectin-1 is the prototype member of the galectin families, existing as a homodimeric 14 kDa soluble protein. Each subunit contains a CRD which is responsible for binding β-galactoside. Numerous N-and O-glycan enriched molecules act as galectin-1 binding targets, including CD2, CD3, CD7, CD43, CD45, CD95, neurophilin-1, lysosome-associated membrane protein (Lamp)-1, laminin and fibronectin [36–38]. Involvement of galectin-1 in cell–cell and cell–extracellular interactions through binding to the various ligand receptor molecules provide multi-functional capabilities, including modulation of cell adhesion, migration, proliferation [27, 28], immunomodulation [39, 40] and angiogenesis [31–33]. These pathologic processes contribute considerably to the generation of the pro-tumorigenic environment, particularly by suppressing anti-tumor T cell-mediated immune responses and promoting tumor angiogenesis.
Previous studies demonstrated that targeting galectin-1 modulates CD4+ and CD8+ T cell-mediated immune responses [22, 32], Treg cell function [25] as well as endothelial cell migration and proliferation [33]. These findings strongly suggest that targeting galectin-1 should promote tumor suppressive effects by enhancing anti-tumor T cell immune responses as well as concurrently suppressing tumor angiogenesis. In this study, we have used the galectin CRD inhibitor, TDG as a synthetic and non-metabolizable disaccharide, which shows relatively high affinity for the galectin-1 CRD (Kd of ~78 μM) [41]. It was shown in B16F10 melanoma and 4T1 breast cancer murine models that intratumoral TDG treatment significantly decreased tumor growth rates with a concomitant increase in CD4+ and CD8+ T cell infiltration into the tumor stroma as well as reduced tumor angiogenesis [32]. Additionally, TDG did not alter tumor growth rates in G1KD B16F10 and 4T1 models, indicating that the main target of TDG and its actions is likely to be via galectin-1-mediated effects. The present study investigated the effects of blocking galectin-1 functions in lung metastasis using TDG or shRNA G1KD. We administered TDG via intravenous injection to reflect a more clinically applicable approach compared to the previous report where intratumoral administration was used. Significantly reduced lung metastases were observed in both 4T1.2 and CT26 murine models following galectin-1 blocking with either TDG or shRNA. The mechanisms by which reduced metastases was achieved through galectin-1 blocking was further explored.
The key immune cells involved in cancer immunology include the Th (CD3+CD4+) type 1 cells and CTLs (CD3+CD8+). Th1 cells help in the activation of CTLs which produce cell lytic molecules such as perforin, granzyme and FasL (CD95L) to kill the target cancer cells [42] underpinning their importance in cancer immunotherapy. The high number of circulating cancer cells correlates with increased metastases and poorer overall survivals [43]. Therefore, the increased levels of peripheral T cell after blocking galectin-1 is proposed to explain one of the mechanisms for the reduced lung metastases.
Furthermore, galectin-1 blocking via TDG treatment or G1KD increased the number of CD3+ T cells as well as the levels of TUNEL+ apoptotic cells in the lung metastatic lesions. This result suggests that targeting galectin-1 not only increases the levels of circulating T cell surveillance, but also affects the immediate T cell responses within the lung metastatic regions. Infiltration of immune cells was significantly associated with reduced metastasis and is consistent with CD8+ CTL infiltration, previously shown to reduce lymphatic and perineural metastatic invasion [3]. It has also been shown that a significant increase in the effector memory T cells (CD8+CD45RO+CCR7−) in primary colorectal tumors correlated with the absence of metastatic invasion [43]. It is known that effector memory T cells reduce O-glycan structures on their surfaces [44], suggesting that memory T cells could thereby escape galectin-1-mediated apoptosis [45]. However, it is not yet known whether increasing the level of Th cells or CTLs by targeting galectin-1 could indirectly affect the population of effector memory T cells to enable to efficiently suppress metastasis.
Previous studies have shown that CD44 and CD326 are responsible for cancer cell attachment to the endothelial cells [13, 14], helping extravasation of metastatic cells. In particular, CD44v is important for cancer cells to extravasate and settle in a second target organ [13]. Since CD44 is normally highly glycosylated in cancer cells [46] and CD326 contains up to three N-glycans per molecule [14], we hypothesized that galectin-1 may interact with CD44 or CD326 on the cancer cells by binding N- or O-glycans expressed on these molecules. The data from the immunoprecipitation and galectin-1 binding assay using CD44low/high or CD326low/high cells revealed that galectin-1 likely binds to CD44 and CD326 expressed on 4T1 breast cancer cells. This would provide a possible mechanism for galectin-1-mediated promotion of cancer cell adhesion. Galectin-1 bound CD44 and CD326 could increase cancer cell affinity to the endothelium in target tissues helping to promote metastatic development. Therefore, targeting galectin-1 would be likely to suppress the initiation of metastasis.
Additionally, CD44 and CD326 are known as markers of CSCs which are defined by their highly tumorigenic and long term self-renewal capacity, enabling regeneration into differentiated cell lineages that contribute to tumor heterogeneity. Given that only one single cell is sufficient to initiate a metastatic lesion [47], it is theoretically possible that CSCs are highly able to initiate metastasis, while the migration of non-CSCs are unlikely to result in tumor disseminate and the formation of heterogeneous metastatic lesions.
As we found that CD44 and CD326 are likely galectin-1 binding targets, galectin-1 may be protecting circulating CSCs from immune surveillance through inducing apoptosis in activated T cells as well as promoting CSC adhesion to endothelium in secondary organs. However, a previous study reported that anti-CD44 mAb did not inhibit galectin-1 binding to T cell surfaces [48]. This may be because of the differences in glycosylation status between CD44 on cancer cells and T cells. Moreover, the engagement of CD44 by the other ligands such as hyaluronan may inhibit galectin-1 binding capacity. Additionally, other studies indicate that the terminal α2,3 sialic acid on N-glycans alter the ligand affinities [49]. In order to identify precisely galectin-1 binding targets, it will be necessary to determine the glycosylation levels on the molecule as well as relation to glycan-modifying enzymes such as glycosyltransferases, which are likely to vary on the different the cell types. Furthermore, it should be noted that TDG reduced the cancer cell aggregation promoted by exogenous galectin-1, and hence, inhibiting galectin-1 function may result in a reduced propensity of cancer cells to clump together and thereby become lodged and embedded within the lung capillaries. This may be one of the mechanisms behind the reduction in lung metastasis development mediated by TDG treatment by preventing cell aggregation and adhesion.
In conclusion, targeting galectin-1 may be able to promote anti-metastatic T cell responses, contributing to the elimination of circulating cancer cells and their ability to form metastases. Furthermore, since galectin-1 binds to the CSC markers, CD44 and CD326, targeting galectin-1 may inhibit CSC binding to the endothelial cells in the vasculature of secondary organs as well as increase sensitivity to T cell-mediated immune responses, preventing metastasis initiation. Taken together galectin-1 may be an effective target to reduce tumor metastasis and since TDG protects effector T cells from galectin-1-induced apoptosis, it is expected that targeting galectin-1 may improve the other immunotherapy efficacies.
References
Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9(4):274–284
Chiang AC, Massague J (2008) Molecular basis of metastasis. N Engl J Med 359(26):2814–2823
Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH (2009) Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29(8):1093–1102
Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, Nieda M (2011) Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer 105(6):778–786
Talmadge JE (2010) Immune cell infiltration of primary and metastatic lesions: mechanisms and clinical impact. Semin Cancer Biol 21(2):131–138
Yang L (2010) TGFbeta, a potent regulator of tumor microenvironment and host immune response, implication for therapy. Curr Mol Med 10(4):374–380
Prendergast GC (2008) Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 27(28):3889–3900
Rabinovich GA (2005) Galectin-1 as a potential cancer target. Br J Cancer 92(7):1188–1192
Kortylewski M, Yu H (2008) Role of Stat3 in suppressing anti-tumor immunity. Curr Opin Immunol 20(2):228–233
Watanabe MA, Oda JM, Amarante MK, Cesar Voltarelli J (2010) Regulatory T cells and breast cancer: implications for immunopathogenesis. Cancer Metastasis Rev 29(4):569–579
Iiizumi M, Mohinta S, Bandyopadhyay S, Watabe K (2007) Tumor-endothelial cell interactions: therapeutic potential. Microvasc Res 74(2–3):114–120
Miles FL, Pruitt FL, van Golen KL, Cooper CR (2008) Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis 25(4):305–324
Klingbeil P, Marhaba R, Jung T, Kirmse R, Ludwig T, Zoller M (2009) CD44 variant isoforms promote metastasis formation by a tumor cell-matrix cross-talk that supports adhesion and apoptosis resistance. Mol Cancer Res 7(2):168–179
Baeuerle PA, Gires O (2007) EpCAM (CD326) finding its role in cancer. Br J Cancer 96(3):417–423
Frank NY, Schatton T, Frank MH (2010) The therapeutic promise of the cancer stem cell concept. J Clin Invest 120(1):41–50
Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M (2008) CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med 8(8):784–804
Li F, Tiede B, Massague J, Kang Y (2007) Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 17(1):3–14
Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K et al (1994) Galectins: a family of animal beta-galactoside-binding lectins. Cell 76(4):597–598
Liu FT, Rabinovich GA (2005) Galectins as modulators of tumour progression. Nat Rev Cancer 5(1):29–41
Yang RY, Rabinovich GA, Liu FT (2008) Galectins: structure, function and therapeutic potential. Expert Rev Mol Med 10:e17
Perillo NL, Pace KE, Seilhamer JJ, Baum LG (1995) Apoptosis of T cells mediated by galectin-1. Nature 378(6558):736–739
Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, Giaccia AJ, Koong AC, Le QT (2011) Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res 71(13):4423–4431
Kovacs-Solyom F, Blasko A, Fajka-Boja R, Katona RL, Vegh L, Novak J, Szebeni GJ, Krenacs L, Uher F, Tubak V, Kiss R, Monostori E (2010) Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol Lett 127(2):108–118
Pace KE, Hahn HP, Pang M, Nguyen JT, Baum LG (2000) CD7 delivers a pro-apoptotic signal during galectin-1-induced T cell death. J Immunol 165(5):2331–2334
Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI (2007) Galectin-1: a key effector of regulation mediated by CD4+CD25+T cells. Blood 109(5):2058–2065
Tinari N, Kuwabara I, Huflejt ME, Shen PF, Iacobelli S, Liu FT (2001) Glycoprotein 90K/MAC-2BP interacts with galectin-1 and mediates galectin-1-induced cell aggregation. Int J Cancer 91(2):167–172
Hittelet A, Legendre H, Nagy N, Bronckart Y, Pector JC, Salmon I, Yeaton P, Gabius HJ, Kiss R, Camby I (2003) Upregulation of galectins-1 and -3 in human colon cancer and their role in regulating cell migration. Int J Cancer 103(3):370–379
Camby I, Belot N, Lefranc F, Sadeghi N, de Launoit Y, Kaltner H, Musette S, Darro F, Danguy A, Salmon I, Gabius HJ, Kiss R (2002) Galectin-1 modulates human glioblastoma cell migration into the brain through modifications to the actin cytoskeleton and levels of expression of small GTPases. J Neuropathol Exp Neurol 61(7):585–596
Clausse N, van den Brule F, Waltregny D, Garnier F, Castronovo V (1999) Galectin-1 expression in prostate tumor-associated capillary endothelial cells is increased by prostate carcinoma cells and modulates heterotypic cell–cell adhesion. Angiogenesis 3(4):317–325
Thijssen VL, Hulsmans S, Griffioen AW (2008) The galectin profile of the endothelium: altered expression and localization in activated and tumor endothelial cells. Am J Pathol 172(2):545–553
Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, Verhofstad N, Nakabeppu Y, Baum LG, Bakkers J, Mayo KH, Poirier F, Griffioen AW (2006) Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci USA 103(43):15975–15980
Ito K, Scott SA, Cutler S, Dong LF, Neuzil J, Blanchard H, Ralph SJ (2011) Thiodigalactoside inhibits murine cancers by concurrently blocking effects of galectin-1 on immune dysregulation, angiogenesis and protection against oxidative stress. Angiogenesis 14(3):293–307
Thijssen VL, Barkan B, Shoji H, Aries IM, Mathieu V, Deltour L, Hackeng TM, Kiss R, Kloog Y, Poirier F, Griffioen AW (2010) Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res 70(15):6216–6224
Stannard KA, Collins PM, Ito K, Sullivan EM, Scott SA, Gabutero E, Darren Grice I, Low P, Nilsson UJ, Leffler H, Blanchard H, Ralph SJ (2010) Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett 299(2):95–110
Camp RL, Scheynius A, Johansson C, Pure E (1993) CD44 is necessary for optimal contact allergic responses but is not required for normal leukocyte extravasation. J Exp Med 178(2):497–507
Barrow H, Rhodes JM, Yu LG (2011) The role of galectins in colorectal cancer progression. Int J Cancer 129(1):1–8
Ohannesian DW, Lotan D, Lotan R (1994) Concomitant increases in galectin-1 and its glycoconjugate ligands (carcinoembryonic antigen, lamp-1, and lamp-2) in cultured human colon carcinoma cells by sodium butyrate. Cancer Res 54(22):5992–6000
Baum LG, Pang M, Perillo NL, Wu T, Delegeane A, Uittenbogaart CH, Fukuda M, Seilhamer JJ (1995) Human thymic epithelial cells express an endogenous lectin, galectin-1, which binds to core 2 O-glycans on thymocytes and T lymphoblastoid cells. J Exp Med 181(3):877–887
Motran CC, Molinder KM, Liu SD, Poirier F, Miceli MC (2008) Galectin-1 functions as a Th2 cytokine that selectively induces Th1 apoptosis and promotes Th2 function. Eur J Immunol 38(11):3015–3027
Chung CD, Patel VP, Moran M, Lewis LA, Miceli MC (2000) Galectin-1 induces partial TCR zeta-chain phosphorylation and antagonizes processive TCR signal transduction. J Immunol 165(7):3722–3729
Delaine T, Cumpstey I, Ingrassia L, Le Mercier M, Okechukwu P, Leffler H, Kiss R, Nilsson UJ (2008) Galectin-inhibitory thiodigalactoside ester derivatives have antimigratory effects in cultured lung and prostate cancer cells. J Med Chem 51(24):8109–8114
de Visser KE, Eichten A, Coussens LM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6(1):24–37
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Galon J (2005) Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353(25):2654–2666
Harrington LE, Galvan M, Baum LG, Altman JD, Ahmed R (2000) Differentiating between memory and effector CD8 T cells by altered expression of cell surface O-glycans. J Exp Med 191(7):1241–1246
Galvan M, Tsuboi S, Fukuda M, Baum LG (2000) Expression of a specific glycosyltransferase enzyme regulates T cell death mediated by galectin-1. J Biol Chem 275(22):16730–16737
Rambaruth ND, Dwek MV (2011) Cell surface glycan-lectin interactions in tumor metastasis. Acta Histochem 113(6):591–600
Fidler IJ, Talmadge JE (1986) Evidence that intravenously derived murine pulmonary melanoma metastases can originate from the expansion of a single tumor cell. Cancer Res 46(10):5167–5171
Pace KE, Lee C, Stewart PL, Baum LG (1999) Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol 163(7):3801–3811
Skelton TP, Zeng C, Nocks A, Stamenkovic I (1998) Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J Cell Biol 140(2):431–446
Acknowledgments
The authors acknowledge Dr Robin Anderson, Peter MacCallum Cancer Centre, for providing us the 4T1.2 mCherry+ cell line. We thank Dr Helen Blanchard, the Institute for Glycomics Griffith University, for contribution to recombinant galectin-1 purification. We also thank Dr. Elwyn Gabutero, Dr Andy Wu and Miss Amanda Clark for the manuscript editing. Finally, we thank Dr Adrian Meedeniya and Dr Cameron Flegg, Griffith Imaging Facility, for technical supports in microscopy.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ito, K., Ralph, S.J. Inhibiting galectin-1 reduces murine lung metastasis with increased CD4+ and CD8+ T cells and reduced cancer cell adherence. Clin Exp Metastasis 29, 561–572 (2012). https://doi.org/10.1007/s10585-012-9471-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-012-9471-7