
Abstract
A characteristic of human gastroenteropancreatic neuroendocrine tumors (GEP-NET) is a minute unobtrusive primary tumor which often cannot be detected by common physical examinations. It therefore remains unidentified until the tumor has spread and space-occupying metastases cause clinical symptoms leading to diagnosis. Cases in which the primary cannot be located are referred to as NET with CUP-syndrome (cancer of unknown primary syndrome). With the help of array-CGH (comparative genomic hybridization, Agilent 105K) and gene expression analysis (Agilent 44K), microdissected primaries and their metastases were compared to identify up- and down-regulated genes which can be used as a marker for tumor progression. In a next analysis step, a hierarchical clustering of 41.078 genes revealed three genes [C-type lectin domain family 13 member A (CD302), peptidylprolyl isomerase containing WD40 repeat (PPWD1) and abhydrolase domain containing 14B (ABHD14B)] which expression levels can categorize the metastases into three groups depending on the localization of their primary. Because cancer therapy is dependent on the localization of the primary, the gene expression level of these three genes are promising markers to unravel the CUP syndrome in NET.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human gastroenteropancreatic neuroendocrine tumors (GEP-NETs) are neoplasms with different cellular and hormonal differentiation which arise from the diffuse endocrine cell system. NET are rare with an incidence of 1–4/100.000. Two-thirds of all NETs are located in the GEP system [1, 2]. According to their location and molecular profile they can be classified into gastrointestinal NETs (GI-NETs) and pancreatic NETs (PNETs) [3, 4]. NETs may functionally be active leading to characteristic clinical syndromes. The carcinoid syndrome is the most frequent and is characterized by flushing, diarrhea and right-sided heart disease. Prognostic factors for NETs are the location of the primary tumor, the clinical stage, the presence of liver metastases and the proliferation rate (Karnofsky Index (KI)-67 Index) [5–7].
Surgical treatment is the gold standard, however an anti-proliferative treatment becomes more and more frequent. The location of primary tumors also determines the type of treatment for NETs. NETs derived from ileum or jejunum respond to long-acting somatostatin analogues and show an antiproliferative response compared with a placebo whereas PNETs may be treated by streptozotocin based chemotherapy [8].
The molecular pathways underlying the tumorigenesis of NETs are poorly understood. Genetic aberrations and instability are critical criteria in tumorigenesis which can be detected by microarray based comparative genomic hybridization (CGH). A multiplicity of deregulated genes in NETs have been detected and described in many studies but histopathological data and a context to clinical behavior is still missing in most of these studies [3, 9–12].
Recently, investigations have discovered strong distinctions in the genetic profile of PNETs and other GI-NETs (e.g. small intestine, gastric, rectal) [3, 12]. Between both types losses on chromosomes 3 and 11 and gains on chromosomes 4, 5, 14, 19 and 20 were observed [13–17]. Different modifications, losses of chromosomal regions 3p, 21q, and 6 and gains on 4, 7, 9q, 14q, and Xq are associated with metastatic events [18].
Previous studies have shown in different tumor entities (e.g. lymphoma, colorectal carcinoma) a higher level of similarity in the gene expression profiles between the primary tumors and their metastases than between primary and environmental normal cell tissue [19]. Recently, a correlation in the similarity of the gene expression profile and the WHO classification was noted in well-differentiated PNET. In further studies the influence of gene expression profiles become more and more important to predict the clinical behavior of NETs [20–22].
Therefore, the aim of the present study was to investigate the DNA aberration pattern and the gene expression profiles of GEP-NETs’ primaries and metastases. Beneath an inconsistent DNA aberration profile we found that the expression level of three genes [C-type lectin domain family 13 member A (CD302), peptidylprolyl isomerase containing WD40 repeat (PPWD1) and abhydrolase domain containing 14B (ABHD14B)] in NET metastases is a characteristic for the origin of primaries. These genes can allocate the primary of the metastases to the stomach, the ileum or the pancreas, respectively. Because therapy is often dependent on the localization of the primary, this result could be a promising diagnostic tool.
Materials and methods
Patients and specimen preparation
23 frozen tissue samples from 17 NET-patients including 17 metastases and six primaries were provided from the Department of General and Visceral Surgery, Zentralklinik Bad Berka, Germany. After surgery the tumor samples were frozen immediately to avoid DNA and RNA degradation and stored by −80°C until preparation. The frozen tissue samples were cut into 10 μm thick slices and put on glass slides. One tumor section was stained with hematoxylin and eosin (HE) and histological characterized through an expert’s survey. For DNA and RNA-isolation only the tumor cells from 10 to 20 unstained tumor sections were micro dissected with the help of the laser-based microscope (Microbeam C; Zeiss) and collected in a reaction tube [23, 24]. Two liver metastases in which tumor cells were present in less than 40% were excluded from the analysis.
Array-CGH
To investigate individual chromosomal imbalances in these tumor samples the high resolution array-CGH platform from Agilent was used. Array-CGH experiment was performed with the Human Genome Microarray Kit 105A according to the manufacturer’s instructions (www.agilent.com, protocol v. 5.0, June 2007). For array-CGH 12 NETs were investigated, these comprised of eight metastases and four primaries. The aim was the identification of consistent aberration pattern for primaries and metastases in which chromosomal regions might harbor genes which are involved in the pathogenesis and progression of NETs.
The patient’s genomic DNA (gDNA) was then isolated from the collected tumor-pieces using QIAmp DNA Mini Kit from Qiagen. Depending on the gender of the patient male or female pooled gDNA was used as reference DNA. One microgram of tumor and reference DNA was digested with the restriction enzymes AluI and RsaI. Then both DNA samples were labeled with a fluorescent dye: Cy5 (tumor-DNA) and Cy3 (reference-DNA) with the Agilent Genomic DNA Enzymatic Labeling Kit Plus. Labeled tumor and reference DNA was purified, measured, combined, denatured, pre-annealed with Cot-1 DNA (Roche) and blocking reagent (Agilent) and then hybridized to the 60-mer length oligonucleotide probes on the 105K microarray-surface. These oligonucleotide probes were immobilized onto specially-prepared glass slides using a proprietary non-contact industrial inkjet printing process (www.Agilent.com). This in situ synthesis process prints 60-mer length oligonucleotide probes, base-by-base, from digital sequence files. After 40 h of hybridization in a rotating oven (Agilent Technologies) at 65°C and 20 rpm the microarrays were washed and scanned at 5 μm resolution with an Agilent G2565CA scanner. The raw data (tiff-images) were extracted with Feature Extraction versus 10.5.1.1. (Agilent Technologies) and analyzed with CGH Analytics v 3.5.14 (Agilent Technologies).
Gene expression analysis
Because the genetic mechanism of tumor progression in NETs is still unclear, the gene expression profile of microdissected primaries and metastases should be compared to discover similarities and differences which can be used as a marker for tumor progression in this tumor entity. Furthermore, the genetic signature of the NETs can help to localize the primary in NET patients with cancer of unknown primary (CUP) syndrome. The One Color 44K microarray-based gene expression analysis was performed with the Human Genome Quick Amp Labeling Kit according to the manufacturer’s instructions (www.agilent.com, protocol v. 5.7, March 2008). For gene expression analysis 22 frozen tumor tissue samples of 16 patients were analyzed: including 16 metastases and six primaries and three pairs of primary and metastases of the same patients. Metastases from 14 patients were used for hierarchical cluster analysis.
The 22 fresh frozen tumor samples were microdissected to separate tumor tissue from normal tissue. RNA was isolated from the tumor tissue pieces using the RNeasy Mini Kit from Qiagen. RNA amount, purity and quality were checked with Agilents bioanalyser. The sample input RNA varies between 200 ng and 1 μg. To amplify the target material and to generate fluorescent cDNA (complimentary DNA) simultaneously the Quick Amp Labeling Kit from Agilent was used. T7 RNA polymerase incorporates cyane 3-labeled CTP. Amplified and labeled cDNA samples were purified with the RNeasy Mini Kit from Qiagen and afterwards the cDNA concentration and cyane 3 dye concentration was measured using the NanoDrop ND-1000 UV–Vis Spectrometer v. 3.2.1.
The cDNA got hybridized to the 60-mer length oligonucleotide probes on the 44K microarray-surface. After 17 h of hybridization in a rotating oven (Agilent Technologies) at 65°C and 10 rpm the microarrays were washed and scanned at 5 μm resolution with an Agilent G2565CA scanner. The raw data (tiff-images) were extracted with Feature Extraction versus 10.5.1.1. (Agilent Technologies) and analyzed with the GeneSpring GX v. 10.0.2 (Agilent Technologies). The hybridization quality was checked for spatial artifacts with QC-Reports. The raw data were normalized individually by the quantile method (and by shift to 75 percentile) and background correction was done by using baseline transformation: median of all samples (median polishing), which results in log 2 scale transformed data. To ensure a Gaussian-like distribution and to control the quality, the data were inspected by using histograms, matrix plots, box-whisker plots, correlation plots, quality control metrics plot and by 3D PCA (principal component analysis) scores. Two samples with too high background signal were excluded from the analysis. The statistical analysis was done with ANOVA, with none post hoc test. For the P value computation the asymptotically method was chosen and for P value correction the Benjamin Hochberg FDR (false discovery rate) was chosen. Fold change analysis was performed on the 5,400 selected genes which had an FDR corrected P value <0.05. The fold change cut off was set to ten. 1,760 genes with a fold change ≥10 were hierarchically clustered on entities and conditions by using Euclidian distance metric and centroid linkage rule. A decision tree with very stringent conditions was generated to find handful genes in the metastases which are equally expressed in their primary. So those genes could significant separate the metastases in three groups depending on the localization of the primary.
Immunohistochemistry
To validate the different expressed genes, which were discovered by gene expression analysis, immunohistochemistry (IHC) was performed on frozen tissue sections with the corresponding antibody [25]. The fixation of the tumor sections was done by incubation in ice cold acetone for 9 min and then dried on air for 30 min. The first step of staining was the blocking with biotin. For this purpose avidin solution was brought onto the tumor sections and incubated for 10 min in a wet chamber at room temperature. After washing with TBS-T buffer the glass slides were incubated for 10 min with biotin-solution and then washed again with TBS-T buffer. One hundred microliter of the 1:1,000 diluted first antibody (e.g. rearranged during transfection (RET)-antibody) was applied to the tumor sections. After 1 h of incubation in a wet chamber by room temperature and three washing steps with TBS-T, the second antibody was brought onto the tumor sections. After 30 min of incubation and washing, streptavidin was applied for further 30 min. To ensure the specificity of the staining a levamisol and chromogen solution was applied. The counterstaining was done with hematoxylin and afterwards the blueing of the nuclei was done with tab water. At the end the tumor sections were covered with glycerol-gelatine. The histological evaluation was done through an expert’s survey.
Results
The genetic profile of metastases and their primaries was investigated to detect new targets of tumor progression, to find new accurate prognostic tools and to adapt medical treatment.
Patient’s characteristics
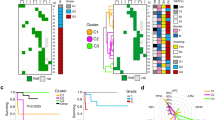
For both analyses a total of 23 frozen tumor tissue samples of 17 patients were analyzed: they comprised of 17 metastases and six primaries and three pairs of primary and metastases of the same patients. Ten of the 17 patients (59%) presented an advanced local spread (T4-status). 15 of the 17 patients (88%) showed lymph node infiltration at the primary. 17 metastases were resected from 14 patients. Ten well differentiated NETs (WDET, KI-67: <5%), two well differentiated endocrine carcinomas (WDEC, KI-67 <10%) and five poorly differentiated carcinomas (PDEC, KI-67: >10%). The median KI was 90% (Table 1). For array-CGH 12 NET were investigated, including eight metastases and four primaries. For gene expression analysis 22 NETs were investigated, including 16 metastases and six primaries. Patient 8 and 27 were NET patients with CUP syndrome and also excluded from cluster analysis. Therefore in total, metastases from 14 patients were used for hierarchical cluster analysis (Fig. 3).
DNA-array-CGH
Each single tumor, independent of primary or metastases, displayed chromosomal imbalances (Fig. 1). No consistent DNA-aberration pattern could be found. In metastases more aberrations could be detected than in primary NET. Small DNA-gains were more frequent than DNA losses. The most frequent gains involved chromosome 16p (42%), 7p (33%), 14q (33%), 15q (33%), 17q (33%). In 25% of all cases 5p, 8q, 9q, and the X-chromosome were amplified. The most common deletions included chromosome 1, 10q, 11, 13q, 14q, 15q, 17p and 21q in 25% of all cases. The whole chromosome 18 was lost in 58% of all tumors. In the chromosomal region 10q11.2 continuous gains or losses were found. This indicates the involvement and a specific function of the RET proto-oncogene in GEP-NET. In order to validate the results concerning the loss and gain of the RET proto-oncogene, these results were exemplarily compared with the results of the gene expression analysis, and IHC was applied on the kryo-sections of the tumors to validate the data on the protein level (Table 3; Fig. 2). For example in the tumor cells of liver metastases 21 a deletion of the RET oncogene could be detected, the gene expression was down-regulated but the IHC shows a negative-moderate staining pattern for RET protein. This IHC result can be explained because the normal liver tissue displayed a moderate staining, not the tumor cells. For array-CGH and gene expression analysis only tumor tissue was used, but IHC was performed on Kryo-sections including tumor tissue and normal cell tissue (Table 3; Fig. 2).

Array-CGH aberration profile of 12 NET samples. The array-CGH aberration profile displays DNA-gains and -losses for each chromosome of 12 investigated NETs
RNA-gene expression analysis and hierarchical clustering can allocate the metastases to their primaries
The gene expression analysis with the 44K microarray from Agilent revealed 22 up-regulated genes and 11 down-regulated in the metastases. All genes exhibit a fold change >10 and an FDR corrected P value <0.05 (Table 2).
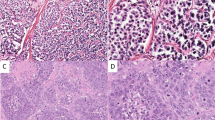
Exemplarily five proteins of these 33 different expressed genes were selected for IHC and thereby the gene expression results could be validated on the protein level. IHC results are exemplarily shown in this paper for RET protein, Cadherin 22 protein, and Dact2 protein whose genes were up-regulated. Furthermore results are shown for interleukin 8 (IL-8) protein, whose gene was down regulated in the metastases (Table 2; Fig. 2). RET, Cadherin 22 and Dact2 were found to be expressed in the cytoplasm and notably strong in the membrane of tumor cells in metastases which were growing invasive into the environmental stroma tissue. Interestingly, the tumor cells in liver metastases were widely not stained for all three proteins, but normal liver tissue displayed a strong reaction. Most metastases were nearly complete negative for IL-8, whereas a strong expression of IL-8 could be detected in tumor cells in the primary. Results are not shown.

IHC for different proteins. a IHC for RET protein on a metastasis in the liver (Table 1, patient 19II). The protein (colored in red) is located in the cytoplasm and/or membrane in the tumor cells. b IHC for Cadherin 2 on a metastasis in the liver (Table 1, patient 4). The protein (colored in red) is located in the cytoplasm and/or membrane in the tumor cells. c IHC for “dapper, antagonist of beta-catenin, homolog 2” (encoded by the DACT2 gene) on a metastases in the stomach (Table 1, patient 7). The protein (colored in red is located in the cytoplasm and/or membrane in the tumor cells. d The metastases of tumor sample 19 consist of negative tumor cells for IL-8 (Table 1, patient 19)
In a next analysis step the gene expression profile of all primaries was separately investigated and throughout a hierarchical cluster analysis three gene clusters including 1,760 different expressed genes could be revealed. These three gene clusters were specific to primaries located in stomach, pancreas and ileum, respectively (Fig. 3). These three cluster caliber (edit format) were applied to the gene expression profiles of all metastases. For each metastasis—independent of where it was located—one of these three gene clusters was almost identical depending where their primary was located.

HeatMap. Hierarchical cluster analysis of 14 neuroendocrine metastases showing the expression profile of 1.760 genes throughout the metastases could be categorized into three groups: “pancreas (n = 6)”, “ileum (n = 6)” and “stomach (n = 2)” depending on the localization of their primary
To make the prediction of the localization of the primary as significant and as specific as possible a statistical decision tree with very stringent conditions was generated and revealed three genes out of these 1.760 which could predict the primary site of origin (Table 4; Fig. 4). The first gene in the decision tree is CD302. If CD302 is up-regulated in the metastasis with a fold change >13 this indicates that the primary is located in the ileum. The next gene in the decision tree is PPWD1. If PPWD1 is down-regulated in the metastasis with a fold change <3 the prediction for the localization of the primary is the pancreas. If ABHD14B is up-regulated with a fold change >4 this indicates that the primary is located in the stomach (Table 4; Fig. 4).

Decision tree. The decision tree illustrates the principle of our three gene classifier to predict the primary site of origin in NETs with CUP syndrome
Those three groups correspond well to the two WHO classifications of GEP-NET categories: GI-NETs (group 1: metastases with primary in the stomach; group 2: metastases with primary in the ileum) and PNETs (group 3: metastases with primary in the pancreas).
The expression level of those three genes (CD302, PPWD1 and ABHD14B) in metastases of two NET patients (8 and 27) with CUP syndrome could give evidence as to where the primary tumor is located.
It can be concluded that in this study a very high level of similarity in the gene expression profile between the primary tumor and their metastases can be shown.
Discussion
NETs are rare and heterogeneous in their clinical outcome. Primary NETs are often difficult to detect and therefore lead to a diagnosis late in their development [2, 26, 27]. NET patients with CUP syndrome cannot benefit from therapies tailored toward distinct tumor types. Due to biological prognostic factors like tumor staging, existence of liver metastases, proliferation rate, whether the tumor is hormone producing or not and most importantly depending on the localization of the primary, the therapy has to be individualized for each NET patient. Therefore for more than 10 years several attempts were made to identify the location of the primary site of origin with specific gene expression signatures of their metastases. It is assumed that in tumor patients with CUP syndrome the metastases have a growth advantage compared to the primary [28]. The underlying genetic mechanism for tumorigenesis and progression is still unclear. It is known that for many solid tumors these mechanisms are caused by chromosomal aberrations which lead either to amplifications of oncogenes or to deletions of tumor suppressor genes. These gains or losses have positive or negative impacts on genome stability, angiogenesis, cell proliferation and apoptosis that are hallmarks of tumorigenesis and metastasis [13, 14, 29, 30]. With the help of genome-wide array-CGH analysis it became relatively easy to detect nearly all copy number variations and loss of heterozygosity (LOH) events with high resolution in frozen and paraffin embedded tumor tissue samples. Beneath these numeric aberrations a changed gene expression is common in malignancies where signal pathways are disturbed and modified. With the help of microarray-based analysis of the gene expression it is possible to illustrate and evaluate the modified profile of all metabolic-, signal- and structural genes, which are necessary for cell proliferation, cell differentiation and apoptosis.
The aim of the present study was to investigate the underlying genetic mechanisms of tumor progression in NET by defining a consistent aberration- and gene expression pattern for primaries and metastases. The second starting point of this study was the investigation of different expressed gene clusters which can be used as an organ specific molecular classifier for neuroendocrine metastases with unknown primary.
Array-CGH
The array-CGH analysis, where we set the cut-off for detected aberrations to 5 Mb, revealed more aberrations in metastases than in primary NETs. This is consistent with a study by Zhao et al. [31]. Overall small DNA gains were more frequent than DNA losses (Fig. 1). Both observations implicate that DNA gains might be associated with higher tumor stages. The most common aberration in the investigated NETs was the loss of the whole chromosome 18 in 58% of the cases. This event was detectable in primaries and metastases which lead to the conclusion that the loss of chromosome 18 paired with further individual aberrations might be an early event in tumorigenesis and tumor progression. This concurs with the conclusions in other publications [32–34]. Interestingly in our study five out of seven tumors with loss of chromosome 18 were classified as WDET with a KI-67 Index <5% (Table 1). Such a low KI-67 Index, however, indicates a slow cell division rate and therefore a slow tumor growth [7]. One explanation for these contradictory results could be that the tumor accumulated so many aberrations that normal mitosis was not possible anymore.
A gain-of-function mutation in the RET proto-oncogene (e.g. DNA amplification) is associated with the development of different types of human cancer including multiple endocrine neoplasia type 2A and 2B and sporadic NETs from different locations (pheochromocytomas, medullar and papillary thyroid carcinomas and parathyroid tumors) [35].
Further genes which were amplified or deleted, respectively, are: BIRC5 (Survivin) MEN1, H19, IGF2, Cyclin D, p53, c-Myc, c-Met, EGFR, erb-B2, E-Cadherin, and wnt which are associated to important cancer pathways or play a central role in cell cycle and tumor progression [3, 32]. It can be concluded that the array-CGH can display an enormous number of DNA aberrations which affect many known proto-oncogenes and tumor suppressor genes. Nevertheless it was not possible to obtain a common aberration pattern for primaries and metastases.
Gene expression analysis
Previous studies on the proteomic level have shown that protein signatures can allocate metastases to a specific tumor entity [19]. It is also known that there is a higher level of similarity in the gene expression profiles between the primary tumors and their metastases than between primary and environmental normal cell tissue [22]. The first three seminal studies about prediction of primaries with the help of microarray based expression profiles of their metastases were published in 2001 by Ramaswamy [36], Su [37] and Yeang [38]. In the entire previously discussed studies microarray based gene expression analysis were used to predict the primary site of origin in patients with CUP syndrome for the most common solid tumor entities but not for NETs. Other groups have successfully shown that gene expression analysis can identify the primary site for CUPs by using databases (e.g. the CupPrint database (www.Agendia.com; Amsterdam, the Netherlands) with multiclass classifier based on different algorithms (e.g. k-nearest neighbor algorithm and vector machine algorithm) [10, 21, 26, 36, 37, 39–51]. Varadhachary et al. [48] evaluated the Veridex assay comprising of 10 genes which allows the localization of the primary in lung, breast, colon, ovary, pancreas and prostate.
There are some studies in which different expressed genes in PNETs are compared to their metastases, compared to a cell lines and compared with other pancreatic tumors to find potentially novel markers for PNETs with CUP syndrome [10, 52, 53]. In other studies researchers compared gene expression patterns in GI-NETs with their metastases and cell lines to find new tumor markers for tumorigenesis [18]. But to our knowledge no studies were published to find different expressed genes between metastases and their primaries of both NET categories (PNET and GI-NETs) paired with a genetic signature for metastases of both categories which may predict the primary site of origin in NET patients with CUP syndrome. Molecular genetic events of both are just described and discussed on the DNA-level discovered by array-CGH [54, 55].
Besides gene expression profiling IHC remains the most important method for phenotypical classification and identification of certain tumor markers in different tissues [56]. Some genes which were discovered by the previous discussed gene expression studies have been established as markers for IHC. In many studies these IHC markers and known histopathological markers were combined to predict the primary site of adenocarcinomas in patients with CUP syndrome [39, 52, 53].
It can be concluded that primaries in breast, colon and lung can be detected easily, primaries located in ileum, stomach, pancreas and biliary tract are more difficult to detect [48, 53]. So these assays (e.g. Veridex) are not useful for NET patients with CUP syndrome when the primary is located in the stomach or ileum which is most often the case. All known classifiers are multiclass classifier which needs at least 10 genes to allocate the primary.
Microdissection
In the field of genomic and proteomic research laser assisted tissue microdissection is an established method to generate homogeneous samples from distinct morphological areas for downstream analyses. Nevertheless, based on citations it is estimated that in array based studies in only about 10% tissue microdissection was used [57, 58]. In our study we used microdissected tumor tissue for both analyses and could therefore establish a decision tree out of three genes (CD302, PPWD1 and ABHD14B) with the help of hierarchical cluster analysis and biostatistical investigations. The three found genes are not directly connected to malignancies. The CD302 protein is a C-type lectin receptor which is involved in cell adhesion and migration, as well as in endocytosis and phagocytosis [59–61]. The PPWD1 gene and its encoding protein peptidylprolyl isomerase containing WD40 repeat is described in just one publication [62]. ABHD14B is almost unknown and no publication could be found.
Their expression level in metastases can predict the primary site of origin for ileum, stomach and pancreas with a prediction score between 0.67 and 1 (Table 4; Fig. 4). The combination of three genes is the smallest described and known classifier to predict the localization of the primary in NET patients with CUP syndrome for both categories (PNETS and GI-NETS). It is obvious that this small classifier of three genes is enabled by microdissection which reduces the complexity of tissue drastically. In all previous studies, larger classifiers were obtained because no microdissection was performed. Recently IHC for these three genes is going on to validate the gene expression results on protein level. Results are not shown.
Conclusion
We developed a stringent classifier including three genes which can predict the site of primary origin in both WHO-classified NET-categories by the use of highly defined tissue material although we analyzed—as all other studies on CUP—a relatively small number of tumor samples. To validate our newly discovered model further blinded and prospective studies have to follow as well IHC experiments.
Abbreviations
- ABHD14B :
-
Abhydrolase domain containing 14B
- CD302 :
-
C-type lectin domain family 13 member A
- CDH22 :
-
Cadherin 22
- DACT2 :
-
Dapper, antagonist of beta-catenin, homolog 2
- CGH:
-
Comparative genomic hybridization
- CUP:
-
Cancer of unknown primary
- GEP-NET:
-
Gastroenteropancreatic neuroendocrine tumor
- GI-NET:
-
Gastrointestinal neuroendocrine tumor
- IHC:
-
Immunohistochemistry
- IL-8:
-
Interleukin 8
- NET:
-
Neuroendocrine tumor
- PDEC:
-
Poorly differentiated carcinoma
- PNET:
-
Pancreatic neuroendocrine tumor
- PPWD1 :
-
Peptidylprolyl isomerase containing WD40 repeat
- RET :
-
Rearranged during transfection (proto-oncogene)
- WDET:
-
Well differentiated neuroendocrine tumor
References
Bornschein J, Kidd M, Malfertheiner P et al (2008) Gastrointestinal neuroendocrine tumors. Dtsch Med Wochenschr 133(28–29):1505–1510
Abbruzzese J, Abbruzzese M, Hess K et al (1994) Unknown primary carcinoma: natural history and prognostic factors in 657 consecutive patients. J Clin Oncol 12(6):1272–1280
Duerr EM, Chung DC (2007) Molecular genetics of neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab 21(1):1–14
Duerr E, Mizukami Y, Ng A et al (2008) Defining molecular classifications and targets in gastroenteropancreatic neuroendocrine tumors through DNA microarray analysis. Endocr Relat Cancer 15(1):243–256
Panzuto F, Nasoni S, Falconi M et al (2005) Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer 12(4):1083–1092
Tomasetti P, Campana D, Piscitelli L et al (2005) Endocrine pancreatic tumors: factors correlated with survival. Ann Oncol 16(11):1806–1810
Scholzen T, Gerdes J (2000) The Ki-67 protein: from the known and the unknown. J Cell Physiol 182(3):311–322
Rinke A, Müller H, Schade-Brittinger C et al (2009) Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 27(28):4656–4663
Berkovic MC, Jokic M, Marout J et al (2007) IL-6-174 C/G polymorphism in the gastroenteropancreatic neuroendocrine tumors (GEP-NETs). Exp Mol Pathol 83(3):474–479
Capurso G, Lattimore S, Crnogorac-Jurcevic T et al (2006) Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr Relat Cancer 13(2):541–558
Hocker M, Wiedenmann B (1998) Molecular mechanisms of enteroendocrine differentiation. Ann N Y Acad Sci 859:160–174
Oberg K (2009) Genetics and molecular pathology of neuroendocrine gastrointestinal and pancreatic tumors (gastroenteropancreatic neuroendocrine tumors). Curr Opin Endocrinol Diabetes Obes 16(1):72–78
Barghorn A, Komminoth P, Bachmann D et al (2001) Deletion at 3p25.3-p23 is frequently encountered in endocrine pancreatic tumours and is associated with metastatic progression. J Pathol 194(4):451–458
Barghorn A, Speel EJ, Farspour B et al (2001) Putative tumor suppressor loci at 6q22 and 6q23–q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol 158(6):1903–1911
Guo SS, Arora C, Shimoide AT et al (2002) Frequent deletion of chromosome 3 in malignant sporadic pancreatic endocrine tumors. Mol Cell Endocrinol 190(1–2):109–114
Guo SS, Wu AY, Sawicki MP (2002) Deletion of chromosome 1, but not mutation of MEN-1, predicts prognosis in sporadic pancreatic endocrine tumors. World J Surg 26(7):843–847
Wang EH, Ebrahimi SA, Wu AY et al (1998) Mutation of the MENIN gene in sporadic pancreatic endocrine tumors. Cancer Res 58(19):4417–4420
Zikusoka MN, Kidd M, Eick G et al (2005) The molecular genetics of gastroenteropancreatic neuroendocrine tumors. Cancer 104(11):2292–2309
Melle C, Ernst G, Schimmel B et al (2008) Colon-derived liver metastasis, colorectal carcinoma, and hepatocellular carcinoma can be discriminated by the Ca(2+)-binding proteins S100A6 and S100A11. PLoS One 3(12):e3767
Modlin IM, Oberg K, Chung DC et al (2008) Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 9(1):61–72
Giordano T, Shedden K, Schwartz D et al (2001) Organ-specific molecular classification of primary lung, colon, and ovarian adenocarcinomas using gene expression profiles. Am J Pathol 159(4):1231–1238
Alizadeh A, Ross D, Perou C et al (2001) Towards a novel classification of human malignancies based on gene expression patterns. J Pathol 195(1):41–52
Melle C, Ernst G, Schimmel B et al (2004) A technical triade for proteomic identification and characterization of cancer biomarkers. Cancer Res 64(12):4099–4104
Melle C, Ernst G, Schimmel B et al (2003) Biomarker discovery and identification in laser microdissected head and neck squamous cell carcinoma with ProteinChip technology, two-dimensional gel electrophoresis, tandem mass spectrometry, and immunohistochemistry. Mol Cell Proteomics 2(7):443–452
Ramos-Vara J (2005) Technical aspects of immunohistochemistry. Vet Pathol 42(4):405–426
Bloom G, Yang I, Boulware D et al (2004) Multi-platform, multi-site, microarray-based human tumor classification. Am J Pathol 164(1):9–16
Abbruzzese J, Abbruzzese M, Lenzi R et al (1995) Analysis of a diagnostic strategy for patients with suspected tumors of unknown origin. J Clin Oncol 13(8):2094–2103
Yuhas J, Pazmiño N (1974) Inhibition of subcutaneously growing line 1 carcinomas due to metastatic spread. Cancer Res 34(8):2005–2010
Albertson D, Collins C, McCormick F et al (2003) Chromosome aberrations in solid tumors. Nat Genet 34(4):369–376
Speel E, Richter J, Moch H et al (1999) Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol 155(6):1787–1794
Zhao J, Moch H, Scheidweiler A et al (2001) Genomic imbalances in the progression of endocrine pancreatic tumors. Genes Chromosomes Cancer 32(4):364–372
Kytölä S, Höög A, Nord B et al (2001) Comparative genomic hybridization identifies loss of 18q22-qter as an early and specific event in tumorigenesis of midgut carcinoids. Am J Pathol 158(5):1803–1808
Löllgen RM, Hessman O, Szabo E et al (2001) Chromosome 18 deletions are common events in classical midgut carcinoid tumors. Int J Cancer 92(6):812–815
Wang GG, Yao JC, Worah S et al (2005) Comparison of genetic alterations in neuroendocrine tumors: frequent loss of chromosome 18 in ileal carcinoid tumors. Mod Pathol 18(8):1079–1087
Komminoth P, Roth J, Muletta-Feurer S et al (1996) RET proto-oncogene point mutations in sporadic neuroendocrine tumors. J Clin Endocrinol Metab 81(6):2041–2046
Ramaswamy S, Tamayo P, Rifkin R et al (2001) Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci U S A 98(26):15149–15154
Su A, Welsh J, Sapinoso L et al (2001) Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res 61(20):7388–7393
Yeang C, Ramaswamy S, Tamayo P et al (2001) Molecular classification of multiple tumor types. Bioinformatics 17(Suppl 1):S316–S322
Horlings H, van Laar R, Kerst J et al (2008) Gene expression profiling to identify the histogenetic origin of metastatic adenocarcinomas of unknown primary. J Clin Oncol 26(27):4435–4441
Dennis J, Vass J, Wit E et al (2002) Identification from public data of molecular markers of adenocarcinoma characteristic of the site of origin. Cancer Res 62(21):5999–6005
Shedden K, Taylor J, Enkemann S et al (2008) Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat Med 14(8):822–827
Buckhaults P, Zhang Z, Chen Y et al (2003) Identifying tumor origin using a gene expression-based classification map. Cancer Res 63(14):4144–4149
Tothill R, Kowalczyk A, Rischin D et al (2005) An expression-based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res 65(10):4031–4040
Khan J, Wei J, Ringnér M et al (2001) Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 7(6):673–679
Talantov D, Baden J, Jatkoe T et al (2006) A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin. J Mol Diagn 8(3):320–329
Dumur C, Lyons-Weiler M, Sciulli C et al (2008) Interlaboratory performance of a microarray-based gene expression test to determine tissue of origin in poorly differentiated and undifferentiated cancers. J Mol Diagn 10(1):67–77
Monzon F, Lyons-Weiler M, Buturovic L et al (2009) Multicenter validation of a 1,550-gene expression profile for identification of tumor tissue of origin. J Clin Oncol 27(15):2503–2508
Varadhachary G, Talantov D, Raber M et al (2008) Molecular profiling of carcinoma of unknown primary and correlation with clinical evaluation. J Clin Oncol 26(27):4442–4448
van Laar R, Ma X, de Jong D et al (2009) Implementation of a novel microarray-based diagnostic test for cancer of unknown primary. Int J Cancer 125(6):1390–1397
Bridgewater J, van Laar R, Floore A et al (2008) Gene expression profiling may improve diagnosis in patients with carcinoma of unknown primary. Br J Cancer 98(8):1425–1430
Ma X, Patel R, Wang X et al (2006) Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch Pathol Lab Med 130(4):465–473
Dennis J, Hvidsten T, Wit E et al (2005) Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res 11(10):3766–3772
Park S, Kim B, Kim J et al (2007) Panels of immunohistochemical markers help determine primary sites of metastatic adenocarcinoma. Arch Pathol Lab Med 131(10):1561–1567
Lubensky I, Zhuang Z (2007) Molecular genetic events in gastrointestinal and pancreatic neuroendocrine tumors. Endocr Pathol 18(3):156–162
Kulke MH, Freed E, Chiang DY et al (2008) High-resolution analysis of genetic alterations in small bowel carcinoid tumors reveals areas of recurrent amplification and loss. Genes Chromosomes Cancer 47(7):591–603
Moll R (2009) The initial CUP situation and CUP syndrome: pathological diagnostics. Pathologe 30(Suppl 2):161–167
von Eggeling F, Ernst G (2007) Microdissected tissue: an underestimated source for biomarker discovery? Biomark Med 1(2):217–219
von Eggeling F, Melle C, Ernst G (2007) Microdissecting the proteome. Proteomics 7(16):2729–2737
Kato M, Khan S, d’Aniello E et al (2007) The novel endocytic and phagocytic C-type lectin receptor DCL-1/CD302 on macrophages is colocalized with F-actin, suggesting a role in cell adhesion and migration. J Immunol 179(9):6052–6063
Kato M, Khan S, Gonzalez N et al (2003) Hodgkin’s lymphoma cell lines express a fusion protein encoded by intergenically spliced mRNA for the multilectin receptor DEC-205 (CD205) and a novel C-type lectin receptor DCL-1. J Biol Chem 278(36):34035–34041
Butler M, Morel A, Jordan W et al (2007) Altered expression and endocytic function of CD205 in human dendritic cells, and detection of a CD205-DCL-1 fusion protein upon dendritic cell maturation. Immunology 120(3):362–371
Davis T, Walker J, Ouyang H et al (2008) The crystal structure of human WD40 repeat-containing peptidylprolyl isomerase (PPWD1). FEBS J 275(9):2283–2295
Disclosure
I have no relevant financial interests related to this manuscript. I certify that all my affiliations with or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript have been disclosed.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nicole Posorski, Daniel Kaemmerer are the authors contributed equally.
Merten Hommann, Ferdinand von Eggeling are the authors contributed equally.
Rights and permissions
About this article
Cite this article
Posorski, N., Kaemmerer, D., Ernst, G. et al. Localization of sporadic neuroendocrine tumors by gene expression analysis of their metastases. Clin Exp Metastasis 28, 637–647 (2011). https://doi.org/10.1007/s10585-011-9397-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-011-9397-5