
Abstract
Microarray expression signature analyses have suggested that extracellular matrix (ECM) gene dysregulation is predictive of metastasis in both mouse mammary tumorigenesis and human breast cancer. We have previously demonstrated that such ECM dysregulation is influenced by hereditary germline-encoded variation. To identify novel metastasis efficiency modifiers, we performed expression QTL (eQTL) mapping in recombinant inbred mice by characterizing genetic loci modulating metastasis-predictive ECM gene expression. Three reproducible eQTLs were observed on chromosomes 7, 17 and 18. Candidate genes were identified by correlation analyses and known associations with metastasis. Seven candidates were identified (Ndn, Pi16, Luc7l, Rrp1b, Brd4, Centd3 and Csf1r). Stable transfection of the highly metastatic Mvt-1 mouse mammary tumor cell line with expression vectors encoding each candidate modulated metastasis-predictive ECM gene expression. Implantation of these cells into mice demonstrated that candidate gene ectopic expression impacts tumor progression. Gene expression analyses facilitated the construction of a transcriptional network that we have termed the ‘Diasporin Pathway’. This pathway contains the seven candidates, as well as metastasis-predictive ECM genes and metastasis suppressors. Brd4 and Rrp1b appear to form a central node within this network, which likely is a consequence of their physical interaction with the metastasis efficiency modifier Sipa1. Furthermore, we demonstrate that the microarray gene expression signatures induced by activation of ECM eQTL genes in the Mvt-1 cell line can be used to accurately predict survival in a human breast cancer cohort. These data imply that the Diasporin Pathway may be an important nexus in tumor progression in both mice and humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The clinical burden of solid tumor dissemination is immense, with most cancer-related deaths a consequence of metastasis. For example, those patients who present with non-disseminated breast cancer have a relatively good prognosis with the ten-year survival of these patients being approximately 80% [1]. However, those with disseminated disease have a much poorer prognosis, with the median survival of patients with metastatic breast cancer at diagnosis estimated to be 2–4 years [1]. The therapeutic advances of recent years have done little to improve this, which has led many to surmise that advanced disseminated breast cancer is an incurable condition [2].
These data misleadingly imply that disease presentation and course in solid cancers are uniform in their natures. This is not the reality in clinical practice however, with substantial variations in outcome frequently observed in patients with seemingly comparable disease patterns at presentation, risk factors and clinicopathological tumor features. There are several explanations as to why this is the case, with one plausible explanation being that current pathological classification systems for primary tumor characteristics do not capture sufficient information for highly accurate prediction of patient outcome. Additional variables may be required to achieve more accurate prognostic indexes. In recent years many investigators have been exploring molecular as well as histopathological markers as additional covariates to consider for prediction of disease outcome. Many reports have now demonstrated that primary tumors with higher propensities to metastasize exhibit characteristic gene expression patterns [3–5], supporting the use of molecular features in clinical prognostic evaluation. There are a number of possible reasons as to why this might be the case, with the most commonly cited being that metastasis-prone tumors develop somatic aberrations early in their evolution that drive these characteristic gene expression patterns [6].
Another plausible and somewhat complementary explanation for the disparate clinical behaviors of tumors with similar pathological features is the modifying effect of hereditary germline-encoded variation upon the disease process. Work in our laboratory using the highly metastatic polyoma middle-T (PyMT) transgenic mouse model has demonstrated the significant influence of germline variation on tumor progression [7, 8]. Tumors induced by the same oncogenic event, activation of the PyMT transgene, on different mouse genetic backgrounds were shown to have significantly different predilections for pulmonary metastasis. These results suggested that metastasis susceptibility was likely to be due not only to somatic events occurring during the evolution of the tumor, but also due to combinations of inherited germline polymorphisms acting in concert to modulate the probability of whether a tumor will progress. Further investigation of these initial observations allowed us to identify two of these polymorphic metastasis susceptibility genes that modulate metastasis in both mouse and human: the Rap-GTPase activating protein (GAP) Sipa1 [9]; and Rrp1b, a gene that modulates the enzymatic activity of Sipa1 [10].
Additional studies have allowed us to examine whether there is likely an intimate relationship between hereditary polymorphism and tumor gene expression patterns, i.e. genetic polymorphism may not only modulate metastasis efficiency, but also may be an important factor in the induction of the prognostic signature profiles [11, 12]. Examination of previously published metastasis-predictive signatures in both the mouse [11, 12] and in humans [3–5] have revealed that dysregulation of extracellular matrix (ECM) gene expression is a common feature of tumors with a higher propensity to metastasize. Indeed, ECM gene dysregulation has been shown to be a very prominent feature of metastatic progression, and may well explain why highly metastatic mouse mammary tumor cell lines are typically more adhesive, invasive, and migratory than the less metastatic lines [13].
AKXD recombinant inbred (RI) mice [14] were used to determine whether this ECM dysregulation was at least partially under germline control. This methodology not only allowed us to identify Rrp1b as a dual ECM and metastasis efficiency modifier, but also to demonstrate that the cell growth regulator Bromodomain 4 (Brd4) [15–17], which also binds Sipa1, has similar properties with regards to metastasis and ECM gene expression (Bromodomain 4 activation predicts breast cancer survival; N. Crawford, J. Alsarraj, L. Lukes, R. Walker, H. Yang, M. Lee, K. Ozato, K. Hunter; in submission). Furthermore, we demonstrated that differential activity of BRD4 in primary breast cancers accurately predicts outcome in five different tumor gene expression datasets.
The current study represents a continuation of this earlier work to characterize novel metastasis efficiency modifier genes. By using techniques similar to those outlined above, we identify five novel candidate dual ECM and metastasis efficiency modifiers. Furthermore, our data demonstrate that these genes are elements of a potential novel transcriptional network, the activation of which is predictive of outcome in human breast cancer.
Materials and methods
Expression QTL mapping
Microarray hybridization methodology and generation of the microarray expression data from AKXD × PyMT primary tumors has been described previously [12]. Affymetrix CEL files were normalized using the RMA method, averaged for each AKXD RI strain, and loaded into the GeneNetwork web service (http://www.genenetwork.org) [18]. GeneNetwork databases were then searched for probe sets within the eleven genes from our previously described metastasis signature profile [12] classified as being an “ECM component” (Col3a1, Fbln2, Fbn1, Mfap5, Mmp2, Mmp16, Nid1, Serpinf1, Serping1, Timp2, and Tnxb). Additionally, we also included probe sets from the two ECM genes (Col1a1 and Col1a2) represented the eleven gene human breast carcinoma metastasis gene signature profile described by Ramaswamy et al. [3].
Cell culture and development of Mvt-1 clonal isolates ectopically expressing candidate genes
The Mvt-1 cell line, which is derived from an explant-cell culture of primary mammary tumors from MMTV-c-Myc/MMTV-Vegf bi-transgenic mice [19], was obtained as a gift from Lalage Wakefield (NCI, Bethesda). These cells were cultured as described [10]. Mammalian expression vectors encoding the full-length cDNAs for all ECM eQTL candidate genes other than Brd4 were obtained from the Mammalian Gene Collection (Supplementary Table 1). A full-length cDNA for Brd4 in the expression vector pFLAG-CMV2 (Sigma-Aldrich, Saint Louis, MO) was obtained as a gift from Keiko Ozato (NICHD, Bethesda). The control cell line was generated using the vector pCMV-SPORT-β-Galactosidase (Invitrogen, Carlsbad, CA). Transfections and clonal isolations were performed as described previously [10]. Colonies were screened by quantitative real-time PCR (qPCR) as described below to identify clones ectopically expressing candidate genes.
qPCR gene expression analysis
Total RNA was isolated from Mvt-1 clonal isolates as described [10]. cDNA was synthesized from RNA isolated from either primary tumor tissues or transfected cell lines using the ThermoScript RT-PCR System (Invitrogen) by following the manufacturer’s protocol. Single RT-PCRs were performed for each Mvt-1 clonal isolate. SYBR Green or TaqMan qPCR was performed to detect the cDNA levels of ECM eQTL candidates and a variety of metastasis-predictive ECM genes (see above) using an ABI PRISM 7500 and/or 7900HT Sequence Detection Systems (Applied Biosystems, Foster City, CA). Primer and fluorogenic probe sequences for chromosome 7, 17 and 18 eQTL candidates and the housekeeping gene Peptidylprolyl Isomerase B (Ppib) are shown in Supplementary Table 2. Primers for metastasis-predictive ECM genes have been described elsewhere [10]. Reactions were performed using QuantiTect SYBR Green Master Mix (Qiagen) or TaqMan Universal PCR Mastermix (Applied Biosystems) per the manufacturer’s protocol. The cDNA level of each eQTL candidate or metastasis-predictive ECM gene was normalized to Ppib cDNA levels using custom-designed primers for SYBR green-quantified target genes or custom-designed primers and fluorogenic probe for TaqMan-quantified target genes.
Spontaneous metastasis assays
One hundred thousand cells stably expressing either as single eQTL candidate or pCMV-SPORT-β-Gal were subcutaneously implanted into six week old virgin FVB/NJ mice as described previously [10]. These experiments were performed in compliance with the National Cancer Institute’s Animal Care and Use Committee guidelines. Animals were then aged for 28 days before being euthanized. Total tumor weight was determined by complete dissection of the primary tumor. Metastases counts were determined by surface pulmonary counts.
Immunoblot analysis
Transfected Mvt-1 cells were trypsinized and collected, and cell pellets lysed in cell lysis buffer [M-PER® Mammalian Protein Extraction Reagent (PIERCE, Rockford, IL) supplemented with Halt Protease Inhibitor cocktail kit (PIERCE, Rockford, IL)]. SDS-PAGE was performed for 60–90 min at 120 V using the XCell SureLock TM Mini-Cell (Invitrogen, CA) with NuPAGE Novex Bis-Tris Gels (Invitrogen, CA). Proteins were transferred to ImmobilonTM-P membranes (Millipore, Bedford, MA) and immunoblotted against KAI1 (SC-1087; Santa Cruz) and NM23-H1 (SC-343; Santa Cruz) to detect the levels of these proteins in response to ectopic expression of ECM eQTL candidate genes. To ensure uniformity in sample loading onto the gel, membranes were re-immunoblotted against GAPDH (Santa Cruz) or β-ACTIN (Abcam).
Microarray and survival analysis
RNA extraction and processing for Affymetrix GeneChip analysis have been described elsewhere. High confidence human transcriptional signatures of individual ECM eQTL candidate gene expression were generated in exactly the same manner as described for Rrp1b in our earlier work [10]. Briefly, Affymetrix microarrays were used to compare gene expression in three to four Mvt-1 clonal isolates for ECM eQTLs and three Mvt-1/β-galactosidase clonal isolates. CEL files were analyzed using the Affymetrix GeneChip Probe Level Data RMA option of BRB ArrayTools 3.5.0. Genes with <1.5 fold-change from the gene’s median value in 50% of samples, or a log-ratio variation P>0.01 were eliminated from analyses. To identify candidate gene expression signatures, the Class Comparison tool of BRB ArrayTools was performed, using a two-sample t-test with random variance univariate test. P-values for significance were computed based on 10,000 random permutations, at a nominal significance level of each univariate test of 0.0001. Tumor gene expression data from a well described breast cancer cohort [5] were downloaded from the Rosetta Company website (http://www.rii.com/publications/2002/vantveer.html).
Survival analysis was performed as described [10]. BRB ArrayTools was used to perform unsupervised clustering based on individual ECM eQTL signature gene expression. Clustering was performed using average linkage, the centered correlation metric and center the genes analytical option. Samples were assigned into two groups based on the first bifurcation of the cluster dendogram, and Kaplan–Meier survival analysis was performed using the Survival module of the software package Statistica. Significance of survival analyses was performed using the Cox F-test.
Results
eQTL mapping in AKXD recombinant inbred strains reveals three loci controlling the expression of metastasis-predictive ECM genes
The initial aim of this study was to define ECM expression quantitative trait loci (eQTLs). Traditionally, QTL analysis has involved defining specific genomic regions linked to a physiological trait (e.g. pulmonary metastasis burden). However, the advent of microarray technology has allowed global gene expression patterns to be treated as a quantifiable trait, which in turn has facilitated the characterization of eQTLs. An eQTL is therefore a genetically defined genomic locus associated with variation of gene expression (e.g. ECM gene expression). We utilized AKXD RI mice [14] for ECM eQTL mapping. RI panels are a specialized genetic mapping resource that has unique properties that make them particularly valuable for the study of complex traits [20, 21]. The AKXD RI mice are a particularly useful tool for the study of germline-encoded metastatic propensity since they are derived from AKR/J, a highly metastatic strain, and DBA/2J, a weakly metastatic strain [7]. Detailed methodology for the generation of ECM eQTLs has been described previously [10].
Briefly, extracellular matrix (ECM) genes are ubiquitous components of metastasis-predictive expression signatures in both human breast tumor tissue [3–5], and in PyMT-induced mouse mammary tumors [11, 12]. Genetic mapping experiments were performed to identify genomic intervals associated with ECM component transcriptional control in our mouse metastasis model system using our previously derived microarray expression analyses of the PyMT-induced primary tumors in the AKXD RI panel [12]. eQTLs were mapped using probe sets from thirteen previously identified metastasis-predictive ECM genes. A number of reproducible loci were evident [10] although the loci on chromosomes 7, 17 and 18 were of most interest due to their highly consistent correlation with expression of metastasis-predictive ECM probe sets. This is best illustrated when ECM eQTLs are mapped with those probe sets most strongly correlated with metastasis susceptibility (11 probe sets from a PyMT-induced metastasis-predictive gene signatures were identified [12], and five probe sets from the 17 gene human breast carcinoma metastasis gene signature profile described by Ramaswamy et al. [3]). Such analyses demonstrate that expression of most of these sixteen metastasis-predictive ECM probe sets are least suggestively correlated with the loci on chromosomes 7, 17 and 18 (Supplementary Table 3).
Further evidence for the role of these chromosomal regions in modulation of metastatic capacity is derived from the observation that all three ECM eQTLs co-localized with suggestive metastasis QTLs observed after composite interval mapping of the AKXD data. We argue that this observation is consistent with the hypothesis that the ECM eQTLs and the metastasis efficiency QTLs might be causally linked. Chromosomal substitution strains have also been generated for chromosomes 7 and 17, both of which were shown to suppress metastatic capacity [22]. Thus, evidence for a potential causal link between metastasis efficiency and ECM gene expression exists for all three chromosomes.
Identification of ECM eQTL candidate genes
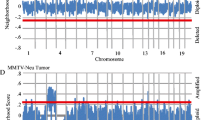
Correlation analysis was performed to identify potential candidates for the chromosomes 7, 17 and 18 ECM modifiers since the current prevailing hypothesis that most modifiers are likely to result from modest variations in gene expression levels or mRNA stability [23, 24]. Whole genome correlation analysis of the microarray data was performed using the Trait Correlation function of the WebQTL database within the GeneNetwork web service [18] to identify probe sets with high correlation coefficients with each of the ECM genes of interest. Probes sets from the whole genome analysis that reside within the candidate eQTL intervals were then examined to identify genes that were reproducibly present in the ECM eQTL gene lists. Twenty-eight genes located within the region of chromosome 7 spanning the peak likelihood ratio statistic (a derivation of LOD score; LRS) score (physical locations ∼56–121 Mb; Fig. 1a) displayed both a high degree of correlation and a low P value with regard to expression of two or more of the 9 probe sets within metastasis-predictive ECM genes (Supplementary Table 4). Regarding the chromosome 18 ECM eQTL (physical location ∼37–76 Mb; Fig. 1b), expression of probe sets representing sixteen genes located within this locus were correlated with the expression of metastasis-predictive ECM probe sets (Supplementary Table 5). The chromosome 17 ECM eQTL has been described in detail elsewhere [10], but thirty candidate genes located within the peak LRS score region (physical locations ∼18–40 Mb) were correlated with metastasis-predictive ECM genes.

Chromosome 7 and 18 ECM eQTLs. ECM eQTL mapping in AKXD RI mice reveals that the expression of metastasis-predictive ECM genes are linked to a number of genomic loci. Analysis of metastasis class-predictive ECM gene component expression patterns in PyMT transgene-induced tumors in AKXD recombinant inbred mice reveals the presence of a number of reproducible eQTLs. ECM eQTL linkage maps were generated with GeneNetwork web service. (a) The physical location of the chromosome 7 eQTL is ∼56–121 Mb. (b) The physical location of the chromosome 18 ECM eQTL is ∼37–76 Mb. The identities of probe sets defining individual ECM eQTLs are given in Supplementary Table 3
Seven genes were observed in the three candidate intervals that demonstrated reproducible associations with ECM gene expression across the AKXD panel (Table 1). Although there are many plausible ECM eQTL candidates within the three eQTL loci (see Supplementary Tables 4 & 5 and Crawford et al. [10]) we chose to further characterize the seven candidates for the following reasons. Three of these genes are of particular interest based on published literatures. Necdin (Ndn) is an imprinted transcription factor on chromosome 7 that has previously been implicated in cellular proliferation, collagen gene expression [30], and regulation of the metastasis-associated gene Hif1a [31, 32]. Brd4, on chromosome 17, is a bromodomain-containing protein that associates with chromatin [17]. Intriguingly, Brd4 has been demonstrated to be binding partner of our recently described metastasis efficiency modifier, Sipa1 [16]. Further characterization of this gene in our laboratory demonstrated that differential expression of Brd4 drives a gene expression signature that predicts outcome in breast cancer (Crawford et al., in submission). Csf1r is a receptor for colony stimulating factor and is important in mammary development, oncogenesis and macrophage recruitment [33]. Studies using knockout mice in the PyMT model have suggested that CSF signaling is an important component for metastatic progression [34]. A fourth gene, Rrp1b, was also considered of great interest since functional genomics studies in our laboratory identified it as potentially binding to the polymorphic PDZ domain of Sipa1 [10]. Furthermore, additional characterization of Rrp1b showed that this gene modulates metastasis efficiency in mice and that the human homolog of Rrp1b contains a polymorphism that is associated with various markers of outcome in breast cancer [10]. The final three genes (Pi16, Luc7l and Centd3) were chosen for further characterization as they possess either a plausible functional role in metastasis (Table 1), a high degree of correlation with metastasis-predictive ECM probe sets (Supplementary Table 5 and Crawford et al. [10]), or a combination of both.
The three reproducible ECM eQTL peaks were then examined to see whether they function in a common transcription regulatory pathway, rather than independently regulating ECM gene expression. Each of the ECM eQTL candidate genes was subjected to eQTL analysis to determine what regions of the genome were associated with steady state mRNA levels (data not shown). Probe sets from each gene in question were examined using the interval mapping function of GeneNetwork to identify genetically defined regions in the AKXD RI panel that were associated with probe intensity on the Affymetrix chips, as a surrogate for steady state gene expression levels. Interestingly, mRNA levels of Ndn, on chromosome 7 appeared to be influenced by the eQTLs on chromosomes 17 and 18, which co-localize with the ECM eQTLs. Analysis of the genes on chromosome 17 revealed the potential influence of the chromosome 18 locus on gene expression of all the candidates in the chromosome 17 locus. The data were therefore consistent with the possibility of a transcriptional cascade, with the chromosome 18 locus modulating expression of candidate genes within the chromosome 17 locus. This locus, in turn, appears to modulate the expression of Ndn on chromosome 7.
Ectopic expression of eQTL candidate genes modulates the expression of metastasis-predictive ECM genes
To evaluate the role that these genes play in the modulation of metastasis-predictive ECM genes and to gain support for the hypothesized transcriptional pathway, transfection experiments were performed. Mammalian expression vectors were obtained from the MGC collection (Supplementary Table 1) and transfected into the FVB/NJ mouse mammary tumor cell line Mvt-1 [19]. Four independent clonal cell lines stably expressing individual ECM eQTL candidate genes as well as control cell lines stably expressing β-galactosidase were isolated by limiting dilution, and qPCR used to confirm candidate gene ectopic expression. Quantitative real-time PCR was also used to determine the expression of transcripts defining the three ECM eQTLs (Col1a1, Col1a2, Col3a1, Fbn1, Mmp2, Nid1, and Serping1; see Supplementary Table 3) as well as five related ECM genes (Col5a3, Col6a2, Fbln2, Mfap5, and Serpinf1). Ectopic expression of each of the candidates was shown to dysregulate the expression of different metastasis-predictive ECM genes (Table 2). The effects of ectopic expression of Rrp1b and Brd4 upon ECM gene transcription have been described elsewhere [10] (Crawford et al., in submission).
Spontaneous metastasis assays demonstrate suppression of tumor growth and progression
Mvt-1 cells ectopically expressing eQTL candidate genes were implanted in the vicinity of the mammary fat pad of FVB/NJ mice to assess the effect of those genes upon tumor growth and metastasis. Following a 28 day incubation period, mice were sacrificed and primary tumor weight was measured and pulmonary surface metastases counted (Table 3). The control cell line data represent the tumor growth and metastatic potential of one Mvt-1/β-galactosidase. One control cell line was used to minimize the number of animals in experiments since we have previously demonstrated all of the control cell lines to be indistinguishable for in vitro and in vivo growth and metastasis characteristics (unpublished data). The effects of ectopic expression of Rrp1b and Brd4 on tumor growth and metastasis in this spontaneous metastasis model have been described elsewhere [10] (Crawford et al., in submission).
Ectopic expression of each of the candidate genes profoundly impacted the tumor growth resulting from subcutaneous implantation of Mvt-1 clones (Table 3). This does not appear to result from a generalized reduction in cellular growth rate since previous growth curve analyses have demonstrated no differences in the in vitro growth characteristics of the ECM eQTL candidate and control cell lines ([10], Crawford et al. in submission and unpublished observations). Furthermore, these data are concurrent with our earlier studies that have demonstrated that ectopic expression of both Rrp1b and Brd4 profoundly reduce tumor growth and metastasis [10] (Crawford et al., in submission).
A significant reduction in secondary lesion formation was also observed in all but the Mvt-1/Pi16 cell lines. However, given that the ECM eQTL candidate genes may be acting as growth suppressors in vivo, it is unclear at this stage whether this reduction in pulmonary metastatic burden is a primary event or secondary to the reduced tumor growth rate. In contrast to all of the other cell lines, the Mvt-1/Pi16 cell line appears to behave disparately: tumor growth is markedly reduced yet the metastatic potential of these cell lines has been maintained. Therefore, the cell lines ectopically expressing Pi16 display a net increase in the rate of secondary lesion implying that Pi16 might actually be a metastasis enhancer, at least in the highly metastatic Mvt-1 cell line. Further investigation will be required to see whether this is the case in other systems.
Microarray and qPCR gene expression analysis demonstrate that eQTL candidates form a metastasis-related transcriptional network
Affymetrix microarrays were used to compare gene expression in three to four Mvt-1 clonal isolates for ECM eQTLs and three Mvt-1/β-galactosidase clonal isolates. Probe sets significantly up- and down-regulated in response to ectopic expression of Ndn, Pi16, Luc7l, Centd3 and Csf1r according to these criteria are listed in Supplementary Tables 6–10, respectively. Microarray expression signatures indicative of activation of Rrp1b and Brd4 in the Mvt-1 cell line have been described elsewhere [10] (Crawford et al., in submission).
ECM eQTL mapping has raised the possibility that candidate genes are components of a transcriptional pathway (see above). The feasibility of such a functional relationship is strengthened when one considers that ectopic expression of each candidate in the Mvt-1 cell line has similar phenotypic effects (e.g. modulation of ECM gene expression, tumor growth and metastasis). If this is actually the case then dysregulation (in the form of ectopic expression) of individual candidate genes should impact the expression of other transcriptional pathway components. Therefore, in addition to the microarray analysis described above, the effect of ectopic expression of each candidate gene on the cellular transcriptional profile of Mvt-1 cells was also analyzed by qPCR (Supplementary Table 11). Additionally, we also used qPCR to analyze the expression of a number of other metastasis-related genes including Tgfb1, the metastasis suppressors Cd82/Kai1 [35] and Tfpi [36] (Supplementary Table 11). A putative transcriptional network was constructed by applying the Occam’s Razor principle, i.e. the simplest explanation is likely correct, by minimizing the number of nodes necessary to explain the results. For example, ectopic expression of Pi16 was shown to up-regulate Brd4 and down-regulate Tfpi, which was also down regulated by ectopic expression of Brd4. The down-regulation of Tfpi by Pi16 could therefore be explained by action through the Brd4 pathway and the putative pathway drawn to represent this potential cascade. We have subsequently termed this putative network the ‘Diasporin Pathway’ based on the potential role in the diaspora, or dissemination, of tumor cells during the metastatic process. Simultaneously, yeast-two hybrid analysis revealed that our previously identified metastasis efficiency gene, Sipa1, putatively interacted with one of the novel chromosome 17 candidate genes (Rrp1b) [10]. Combining these data suggested that Sipa1 and Rrp1b, plus Brd4 which had previously been identified as a Sipa1-interacting partner [16], play a key role in the transcriptional regulation of a number of metastasis-associated genes, including several metastasis suppressors (Fig. 2).

The Diasporin Pathway. A putative transcriptional network that we have termed the “Diasporin Pathway” was constructed by applying the Occam’s Razor principle, i.e. the simplest explanation is likely correct, by minimizing the number of potential interactions that would still explain the results. The pathway consists primarily of the seven ECM eQTL/metastasis efficiency modifier candidate genes as well as a number of factors that are known to be modulators of metastasis. The transcriptional relationship between each gene was determined by using qPCR and microarray expression analysis of the highly metastatic Mvt-1 cell line stably transfected with one of the seven candidate genes. At this stage of analysis, Brd4 and Rrp1b appear to be at the heart of this network since both physically interact with the previously described metastasis efficiency modifier Sipa1
Immunoblot analysis of Mvt-1 clonal isolates
As a means of confirming the downstream effects of ECM eQTL candidate dysregulation as predicted by the Diasporin Pathway, we performed immunoblot analysis to quantify the protein levels of the metastasis-associated factors KAI1 [35] and NM23-H1 [37]. KAI1 is a well characterized factor that suppresses tumor metastasis primarily by inhibiting cancer cell motility and invasiveness [38]. Both microarray and qPCR gene expression analyses suggested that ectopic expression of Diasporin Pathway components should enhance the expression of KAI1. Western blot analysis demonstrated, as expected, that Rrp1b expression significantly altered KAI1 levels (Fig. 3a). Ectopic expression of most of the other Diasporin pathway members also had similar effects which may reflect effects of changing the relative ratios of BRD4 and SIPA1 which previously has been shown to have major impact on cell cycle and cell physiology [16]. Similarly, microarray analysis of Mvt-1 clonal isolates suggested that ECM eQTL candidate dysregulation should suppress expression of NM23-H1. Consistent with these observations, we did observe suppression of NM23-H1 in Rrp1b and Brd4 clonal isolates (Fig. 3b). Convincing evidence of protein alteration in the other cell lines was not observed, probably due the lack of sensitivity of this method to subtle changes in protein levels.

Immunoblot analysis of metastasis-related genes in Mvt-1 clonal isolates. Immunoblot analysis of different ECM eQTL clonal isolates was performed to quantify the effects of candidate gene dysregulation on downstream elements of the Diasporin Pathway. The protein levels of the metastasis-related factors (a) KAI1 and (b) NM23-H1 were quantified. Each lane represents a different Mvt-1 clonal isolate
Diasporin pathway microarray gene expression signatures predict outcome in a well characterized Dutch breast cancer population
Our hypothesis that ECM eQTLs and metastasis susceptibility genes are one and the same leads to the further postulate that the transcriptional signature induced by ectopic expression of these putative dual function genes should induce gene expression signatures that are indicative of both ECM modulation and metastasis. This, in turn, leads to the possibility that such gene expression signatures could be used to predict survival in breast cancer. To evaluate this possibility, the Dutch breast cancer data of van de Vijver [5] were downloaded and the expression signatures induced by candidate gene ectopic expression mapped to the annotations of the Hu25K custom chip. The resulting gene lists were then used to perform unsupervised clustering of human breast cancer microarray datasets. Kaplan–Meier survival analysis was subsequently performed, comparing the survival of the two major clusters formed by the first bifurcation of the dendogram representing high and low levels of candidate gene activation in primary tumor samples (Fig. 4).

ECM eQTL gene expression signatures predict survival in human breast cancer. Diasporin Pathway candidate genes accurately predict overall survival in the Dutch Rosetta dataset. (a) The cumulative survival for the NDN signature was estimated to be 70% vs. 57% for the good and poor prognosis NDN signatures, respectively (NDN signature hazard ratio = 1.94, 95% confidence interval [CI] = 1.16–3.26). (b) The cumulative survival for the LUC7L signature was estimated to be 67% vs. 53% for the good and poor prognosis LUC7L signatures, respectively (LUC7L signature hazard ratio = 1.55, 95% CI = 1.32–2.89). (c) The cumulative survival for the PI16 signature was estimated to be 72% vs. 53% for the good and poor prognosis PI16 signatures, respectively (PI16 signature hazard ratio = 1.57, 95% CI = 1.04–2.36). (d) The cumulative survival for the CENTD3 signature was estimated to be 74% vs. 50% for the good and poor prognosis CENTD3 signatures, respectively (CENTD3 signature hazard ratio = 2.77, 95% CI = 1.80–4.25). (e) The cumulative survival for the CSF1R signature was estimated to be 69% vs. 51% for the good and poor prognosis CSF1R signatures, respectively (CSF1R signature hazard ratio = 2.38, 95% CI = 1.58–3.60). (f) Indeed, it appears that these candidate gene signatures have a similar ability to predict survival in this dataset than the 70-gene signature described by van’t Veer et al. [4]. Specifically, the survival for the good and poor prognosis 70-gene signatures was estimated to be 73% vs. 47%, respectively (70 gene signature hazard ratio = 4.49, 95% CI = 2.65–7.61)
Significant survival differences were observed for those gene expression signatures induced by activation of NDN (Fig. 4a), LUC7L (Fig. 4b), PI16 (Fig. 4c), CENTD3 (Fig. 4d), and CSF1R (Fig. 4e) in the Mvt-1 cell line. Additionally, we have previously demonstrated that the Mvt-1/Rrp1b and Mvt-1/Brd4 gene signatures have also been shown to predict outcome in the same breast cancer cohort [10] (Crawford et al., in submission). This implies that the level of Diasporin Pathway candidate gene activation or candidate gene-associated pathways within a tumor, presumably because of either somatic mutation or germline polymorphism, is an important determinant of the overall likelihood of relapse and/or survival. Further analysis indicated that the differential survival of breast cancer patients was associated with the effects of a distinct subset of genes induced by candidate activation (Supplementary Table 12). Survival analysis using the original 70-gene signature described by van’t Veer and colleagues [5] is shown for comparative purposes in Fig. 4f.
Discussion
We have used a multi-faceted experimental approach to identify a novel transcriptional nexus that appears to be involved in modulation of both ECM gene expression and tumor progression. We initially sought to map ECM eQTLs in AKXD recombinant inbred mice in order to identify those genomic regions where transcript expression displayed a significant degree of correlation with that of metastasis-predictive ECM genes. Our hypothesis here is that the aberrant ECM gene expression observed in tumors more prone to dissemination in both humans [3–5] and mice [11] is to some degree influenced by host germline polymorphism. We identified three loci on chromosomes 7, 17 and 18 that appear to be linked to metastasis-predictive ECM gene expression. Subsequently, correlation analysis was performed to facilitate candidate gene identification since the current prevailing hypothesis is that most modifiers are likely to result from modest variations in gene expression levels or mRNA stability [23, 24]. This revealed a correlation between expression of a variety of genes within the peak region of linkage of each eQTL and the expression of various metastasis-predictive ECM genes (Supplementary Tables 4 & 5 and Crawford et al. [10]). Seven genes were chosen for further analysis, for the reasons outlined above. We must state, however, that differential functionality of these seven candidate genes within AKXD RI mice is highly unlikely to account for all of the linkage observed with the three ECM eQTLs. Indeed, additional plausible candidate genes are evident within the peak regions of ECM eQTL linkage (Supplementary Tables 4 & 5 and Crawford et al. [10]), and it may well be that these too have an effect upon expression of metastasis-predictive ECM genes as well as tumor progression. The aim of this study was not to perform an exhaustive analysis of all factors controlling metastasis-predictive ECM gene expression in AKXD mice, but to identify high-priority metastasis modifier candidate genes. These genes would then be used as a framework upon which we can build a more comprehensive picture of this ECM and metastasis-related transcriptional network.
This approach facilitated the identification of seven high priority ECM and metastasis modulator candidate genes. Although these genes have diverse cellular functions and localizations, our ECM eQTL data suggested that they may well share some form of functional relationship by virtue of their co-regulation with metastasis-predictive ECM genes. Indeed, ectopic expression of each putative ECM eQTL modifier in the Mvt-1 cell line proved that candidate activation not only modulates the expression of metastasis-predictive ECM genes, but also the expression of other members of the assumed transcriptional network. More significantly, we demonstrate that activation of each of these genes in the Mvt-1 cell line suppresses the inherent tumorigenicity of this highly aggressive cell line, with quite profound suppression of tumor growth in some instances (Table 3). Furthermore, we demonstrate that activation of all candidates other than Pi16 suppress metastasis, although we cannot say at present whether this is independent of the reduction of tumor growth rate. Interestingly, it appears that activation of Pi16 in the Mvt-1 cell line reduces tumor growth yet has no overall effect upon the ability of these cells to form secondary lesions. This implies that Pi16 activation in this cell line has facilitated a net increase in the metastatic capacity. Therefore, it would appear at this point that Pi16, unlike the other ECM eQTL candidates, is a tumor suppressor and a metastasis enhancer. Future experimentation will investigate the effects of Pi16 activation in a less aggressive cell line to see whether this observation can be replicated.
However, in spite of these encouraging initial data, the precise relationships between these genes, especially in the broader physiological sense, remain unclear. What is apparent is that the Brd4-Sipa1-Rrp1b relationship seems to be a critical node in this ECM/tumor progression transcriptional network. SIPA1 is a potent modulator of metastatic efficiency in mice, which may relate to its RAPGAP activity [9]. Furthermore, we have demonstrated that SIPA1 is a tumor progression susceptibility gene in humans [39]. Short-hairpin RNA-mediated knockdown of Sipa1 in Mvt-1 cells dysregulates the expression of a variety of metastasis-predictive ECM genes on microarray gene expression analysis (unpublished observations). A plausible mechanism for this ECM dysregulation is that SIPA1 physically interacts with two ECM eQTL candidates identified in this study: BRD4 [16] and RRP1B [10]. Furthermore, physical interactions between SIPA1 and each of these proteins have opposite effects upon the RapGAP enzymatic activity of SIPA1. Specifically, BRD4 increases the RAPGAP activity of SIPA1 [16], whereas RRP1B reduces its activity levels [10]. The significance of these divergent effects upon SIPA1 is particularly intriguing, especially as both Brd4 and Rrp1b appear to have similar effects upon tumor progression. RAP1 activity has been shown to play an important role in metastasis [40, 41]. Therefore, it appears crucial at this stage that we further investigate the BRD4-SIPA1-RRP1B relationship and the influence that it has upon RAP1 levels within the tumor cell to determine whether the effects of these factors are dependent or independent of this system.
The power and potential clinical relevance of this study becomes apparent when one considers that activation of each of Diasporin Pathway components induces a gene expression signature that can be used to predict outcome in human breast cancer. The approach that we have taken is somewhat similar to that of Bild et al. [42], who showed that gene expression signatures can be identified that reflect the activation status of several oncogenic pathways. Specifically, this group demonstrated that in vitro adenoviral-mediated activation of well characterized oncogenic pathways can induce gene expression signatures that predict outcome in a number of different human cancers. We too demonstrate that observations taken from an in vitro setting can be used with effectiveness in a clinical setting. We do, however acknowledge that this approach does have limitations, the most notable of which is that we have only demonstrated the effectiveness of Diasporin Pathway gene expression signatures in one breast cancer cohort. Future efforts in this regards will concentrate upon determining whether the expression signatures identified here hold predictive value in other breast cancer populations as well as in different cancer sub-types. Nevertheless, if these expression signatures do hold substantial predictive value in multiple patient cohorts then it implies that they could enable clinicians to tailor the treatment of individual patients. Specifically, such tumor gene expression signatures could prove useful in prospectively identifying those patients with a seemingly low risk of relapse at presentation based on traditional clinical variables (e.g. node-negative/ER-positive patients) who are likely to eventually relapse. This in turn could facilitate initiation of adjuvant therapy that would not be administered under current treatment protocols. Indeed, the utility of tumor expression profiling is currently being investigated in clinical trials (e.g. TAILORx, MINDACT [43]). We believe that further studies like the current work that reveal novel expression signatures not only serve to enhance our understanding of human breast cancer progression, but more importantly hold the promise of improving upon assessment of prognosis at the time of presentation.
References
Chung CT, Carlson RW (2003) Goals and objectives in the management of metastatic breast cancer. Oncologist 8:514–520
Guarneri V, Conte PF (2004) The curability of breast cancer and the treatment of advanced disease. Eur J Nucl Med Mol Imaging 31(Suppl 1):S149–S161
Ramaswamy S, Ross KN, Lander ES et al (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33:49–54
van’t Veer LJ, Dai H, van de Vijver MJ et al (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530–536
van de Vijver MJ, He YD, van’t Veer LJ et al (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347:1999–2009
Weigelt B, Peterse JL, van’t Veer LJ (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5:591–602
Lifsted T, Le Voyer T, Williams M et al (1998) Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer 77:640–644
Guy CT, Cardiff RD, Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12:954–961
Park YG, Zhao X, Lesueur F et al (2005) Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet 37:1055–1062
Crawford NP, Qian X, Ziogas A et al (2007) Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet 3:e214
Yang H, Rouse J, Lukes L et al (2004) Caffeine suppresses metastasis in a transgenic mouse model: a prototype molecule for prophylaxis of metastasis. Clin Exp Metastasis 21:719–735
Yang H, Crawford N, Lukes L et al (2005) metastasis predictive signature profiles pre-exist in normal tissues. Clin Exp Metastasis 22:593–603
Eckhardt BL, Parker BS, van Laar RK et al (2005) Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res 3:1–13
Mucenski ML, Taylor BA, Jenkins NA et al (1986) AKXD recombinant inbred strains: models for studying the molecular genetic basis of murine lymphomas. Mol Cell Biol 6:4236–4243
Jang MK, Mochizuki K, Zhou M et al (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19:523–534
Farina A, Hattori M, Qin J et al (2004) Bromodomain protein Brd4 binds to GTPase-activating SPA-1, modulating its activity and subcellular localization. Mol Cell Biol 24:9059–9069
Dey A, Chitsaz F, Abbasi A et al (2003) The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci U S A 100:8758–8763
Wang J, Williams RW, Manly KF (2003) WebQTL: web-based complex trait analysis. Neuroinformatics 1:299–308
Pei XF, Noble MS, Davoli MA et al (2004) Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In vitro Cell Dev Biol Anim 40:14–21
Bailey D (1981) Recombinant inbred strains and bilineal congenic strains. In: Fox J (ed) The mouse in biomedical research, 1st edn. Academic, New York
Threadgill DW, Hunter KW, Zou F et al (2004) Genetic modifiers. In: Holland EC (ed) Mouse models of human cancer, 1st edn. Wiley-Liss, New York
Lancaster M, Rouse J, Hunter KW (2005) Modifiers of mammary tumor progression and metastasis on mouse chromosomes 7, 9, and 17. Mamm Genome 16:120–126
Schadt EE, Lamb J, Yang X et al (2005) An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 37:710–717
Chesler EJ, Lu L, Shou S et al (2005) Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet 37:233–242
Maruyama K, Usami M, Aizawa T et al (1991) A novel brain-specific mRNA encoding nuclear protein (necdin) expressed in neurally differentiated embryonal carcinoma cells. Biochem Biophys Res Commun 178:291–296
Kimura E, Hidaka K, Kida Y et al (2004) Serine-arginine-rich nuclear protein Luc7l regulates myogenesis in mice. Gene 341:41–47
Reeves JR, Xuan JW, Arfanis K et al (2005) Identification, purification and characterization of a novel human blood protein with binding affinity for prostate secretory protein of 94 amino acids. Biochem J 385:105–114
Stacey TTI, Nie Z, Stewart A et al (2004) ARAP3 is transiently tyrosine phosphorylated in cells attaching to fibronectin and inhibits cell spreading in a RhoGAP-dependent manner. J Cell Sci 117:6071–6084
Rohde CM, Schrum J, Lee AW (2004) A juxtamembrane tyrosine in the colony stimulating factor-1 receptor regulates ligand-induced Src association, receptor kinase function, and down-regulation. J Biol Chem 279:43448–43461
Hu B, Wang S, Zhang Y et al (2003) A nuclear target for interleukin-1alpha: interaction with the growth suppressor necdin modulates proliferation and collagen expression. Proc Natl Acad Sci U S A 100:10008–10013
Moon HE, Ahn MY, Park JA et al (2005) Negative regulation of hypoxia inducible factor-1alpha by necdin. FEBS Lett 579:3797–3801
Semenza GL (2000) Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol 35:71–103
Wrobel CN, Debnath J, Lin E et al (2004) Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J Cell Biol 165:263–273
Lin EY, Nguyen AV, Russell RG et al (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193:727–740
Jackson P, Marreiros A, Russell PJ (2005) KAI1 tetraspanin and metastasis suppressor. Int J Biochem Cell Biol 37:530–534
Amirkhosravi A, Meyer T, Chang JY et al (2002) Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb Haemost 87:930–936
Salerno M, Ouatas T, Palmieri D et al (2003) Inhibition of signal transduction by the nm23 metastasis suppressor: possible mechanisms. Clin Exp Metastasis 20:3–10
Liu WM, Zhang XA (2006) KAI1/CD82, a tumor metastasis suppressor. Cancer Lett 240:183–194
Crawford NP, Ziogas A, Peel DJ et al (2006) Polymorphisms of SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res 8:R16
Itoh M, Nelson CM, Myers CA et al (2007) Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Cancer Res 67:4759–4766
Retta SF, Balzac F, Avolio M (2006) Rap1: a turnabout for the crosstalk between cadherins and integrins. Eur J Cell Biol 85:283–293
Bild AH, Yao G, Chang JT et al (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439:353–357
Bogaerts J, Cardoso F, Buyse M et al (2006) Gene signature evaluation as a prognostic tool: challenges in the design of the MINDACT trial. Nat Clin Pract Oncol 3:540–551
Acknowledgements
We thank Drs. Doug Lowy and Jude Alsarraj for critical comments on this manuscript. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Crawford, N.P.S., Walker, R.C., Lukes, L. et al. The Diasporin Pathway: a tumor progression-related transcriptional network that predicts breast cancer survival. Clin Exp Metastasis 25, 357–369 (2008). https://doi.org/10.1007/s10585-008-9146-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-008-9146-6